Article Text

Abstract
Background and objectives Chronic neuropathic pain is a common challenging condition following amputation. Recent research demonstrated the feasibility of percutaneously implanting fine-wire coiled peripheral nerve stimulation (PNS) leads in proximity to the sciatic and femoral nerves for postamputation pain. A multicenter, double-blinded, randomized, placebo-controlled study collected data on the safety and effectiveness of percutaneous PNS for chronic neuropathic pain following amputation.
Methods Twenty-eight lower extremity amputees with postamputation pain were enrolled. Subjects underwent ultrasound-guided implantation of percutaneous PNS leads and were randomized to receive PNS or placebo for 4 weeks. The placebo group then crossed over and all subjects received PNS for four additional weeks. The primary efficacy endpoint evaluated the proportion of subjects reporting ≥50% pain reduction during weeks 1–4.
Results A significantly greater proportion of subjects receiving PNS (n=7/12, 58%, p=0.037) demonstrated ≥50% reductions in average postamputation pain during weeks 1–4 compared with subjects receiving placebo (n=2/14, 14%). Two subjects were excluded from efficacy analysis due to eligibility changes. Significantly greater proportions of PNS subjects also reported ≥50% reductions in pain (n=8/12, 67%, p=0.014) and pain interference (n=8/10, 80%, p=0.003) after 8 weeks of therapy compared with subjects receiving placebo (pain: n=2/14, 14%; pain interference: n=2/13, 15%). Prospective follow-up is ongoing; four of five PNS subjects who have completed 12-month follow-up to date reported ≥50% pain relief.
Conclusions This work demonstrates that percutaneous PNS therapy may provide enduring clinically significant pain relief and improve disability in patients with chronic neuropathic postamputation pain.
Trial registration number NCT01996254.
- peripheral nerve stimulation
- neuropathic pain
- phantom pain
- post-amputation pain
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, an indication of whether changes were made, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Chronic neuropathic pain is a common and challenging condition following amputation. There are approximately two million amputees in the USA, with nearly 200 000 amputations performed annually.1 Postamputation pain includes residual limb pain (RLP) and phantom limb pain (PLP), each having significant neuropathic components.2 3 The prevalence of persistent pain has been reported as high as 74% for RLP and 85% for PLP, and many amputees report a combination of RLP and PLP.3 Chronic neuropathic pain in amputees can also decrease function and quality of life, leading to significantly increased risk of depression.4 5
Neuropathic pain in amputees has historically been a complex and challenging condition to treat. Many therapies have been employed, including opioid and non-opioid oral analgesics, nerve blocks, spinal cord stimulation (SCS), and physical and psychological therapies, but few controlled trials demonstrate consistent and effective pain management.2 3 6 7 Peripheral nerve stimulation (PNS) has been used effectively for neuropathic pain, including in cases of postamputation pain.8–10 Historically, PNS leads were designed to be located near or in contact with nerves, often using devices adapted from SCS. This practice was consequently limited by the complexity, cost, and invasiveness of surgical lead implantation, and risk of nerve damage, lead migration, or failure.11–13
Unmet treatment needs for dedicated peripheral neuromodulation applications have resulted in the introduction of new devices and techniques that allow for percutaneous, ultrasound-guided lead implantation10 14 and the development of a PNS system designed specifically for use in the periphery to overcome many of the challenges associated with previous techniques.15 16 A PNS system whose leads may be implanted under ultrasound guidance to provide pain relief for chronic and acute pain indications is now Food and Drug Administration-cleared. The system uses a percutaneous fine-wire coiled lead designed to reduce lead migration and has been reported to have a significantly lower risk of infection than other neurostimulation electrodes.17–21 The availability of such a system provides additional therapeutic options for the treatment of pain.
A previous study demonstrated the feasibility of percutaneously implanting fine-wire coiled PNS leads in proximity to the sciatic and femoral nerves in amputees with neuropathic pain.18 In a series of 16 subjects, 14 (88%) obtained clinically significant relief of RLP and/or PLP. Nine subjects who proceeded to a 2-week stimulation home trial reported an average 72% reduction in RLP and 81% reduction in PLP, and improvements in functional measures like pain interference (81%–83% reductions in RLP and PLP interference).18 Due to the need for data from a randomized study to validate and quantify potential benefits, the present multicenter, randomized, double-blinded, placebo-controlled, partial-crossover study was conducted to collect data on the safety and effectiveness of percutaneous PNS for chronic neuropathic pain in amputees. The primary hypothesis was that percutaneous PNS provides clinically significant pain relief in a statistically significantly greater proportion of subjects than the placebo control group.
Methods
Study design and population
This multicenter, randomized, double-blinded, placebo-controlled, partial-crossover study was designed to collect data regarding the safety and effectiveness of percutaneous PNS for chronic neuropathic pain in amputees. The study was prospectively registered on ClinicalTrials.gov in November 2013.
Consenting patients were assessed for eligibility and randomized at six enrolling sites in the USA. Key inclusion criteria were traumatic lower extremity amputation with healed and healthy residual limb, RLP and/or PLP with an average daily score ≥4 on a 0–10 pain rating scale, and age ≥18 years. Other than requiring a healed and healthy residual limb, there were no constraints on time since amputation. Key exclusion criteria were changes in pain medications in the previous 4 weeks, Beck Depression Inventory-II (BDI-II) score >20, compromised immune system, diabetes mellitus type I or II, implanted electrical stimulation device, history of bleeding disorder or anticoagulation therapy (aside from aspirin or warfarin), history of valvular heart disease, pregnancy, confounding central nervous system (CNS) disorder, allergy to local anesthetic agents or skin-contact materials, history of recurrent skin infection, botulinum toxin injection in the previous 3 months in the affected limb, and steroid injection in the previous 6 weeks in the affected limb. Subjects using warfarin were required to have an International Normalized Ratio (INR) ≤1.5 at the time of implantation. Subjects were permitted to continue use of all existing pain medications during the course of the study. Subjects were permitted to maintain or reduce dosages of pain medications but were asked to not increase dosages above documented baseline levels.
After assessment of eligibility, including completion of a 7-day baseline pain diary to determine average daily RLP and PLP scores and document baseline dosage of pain medications, subjects were randomized 1:1 in blocks of two to a percutaneous PNS therapy group or a placebo control group, stratified by enrolling institution, using a masked allocation sequence generated by the study’s data capture system. Subjects and outcomes assessors were masked to group assignment, and treating physicians were unmasked to allow them to administer interventions accordingly. The PNS group received active stimulation for 4 weeks during the primary outcome evaluation period, while the placebo control group received sham stimulation. After the first 4 weeks, the PNS group received four additional weeks of stimulation (up to 60 days total) and the placebo group crossed over to receive active stimulation for 4 weeks. Interventions ended at the end of the 8-week therapy period, and both groups were followed monthly for up to 12 months from the time of implantation.
Interventions
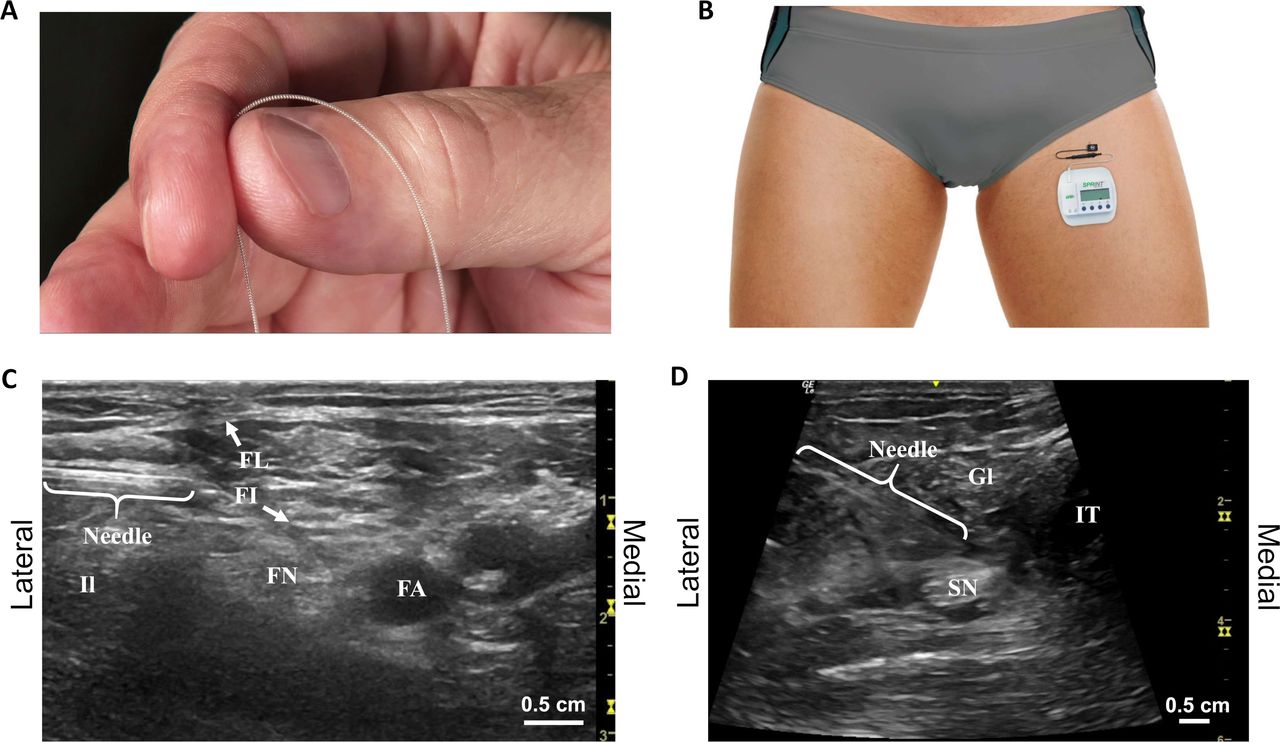
The femoral and sciatic nerves were targeted with percutaneous PNS leads under ultrasound guidance (figure 1).18 A percutaneous lead was directed toward the femoral nerve with the subject supine using a lateral approach approximately 1–2 cm distal to the inguinal crease. The femoral artery, fascia iliaca, and fascia lata were identified as ultrasonic landmarks. The lead was implanted remote (0.5–3 cm) from the nerve to enable selective activation of large-diameter sensory fibers.15 16 18 A lead was also directed toward the sciatic nerve with the subject prone or in lateral decubitus position. Ultrasonic landmarks included the greater trochanter, ischial tuberosity, femur, and/or popliteal artery to guide location of the lead remote from the nerve proximal to the level of amputation. Modifications to these approaches were made at the discretion of the investigator based on patient-specific anatomy.

(A) Fine-wire coiled percutaneous peripheral nerve stimulation leads were implanted and (B) connected to external, body-mounted stimulators. A stimulating needle was used to identify the optimal lead location remote from the targeted (C) femoral and (D) sciatic nerves. FA, femoral artery; FI, fascia iliaca; FL, fascia lata; FN, femoral nerve; Gl, gluteus; Il, iliopsoas; IT, ischial tuberosity; SN, sciatic nerve.
The optimal lead location was determined using a stimulation testing protocol. The insertion site was cleansed using aseptic technique, and cutaneous local anesthesia was administered, taking care to not deliver anesthetic to the target nerve where it may affect the stimulation response. A monopolar needle electrode was inserted to within 0.5–3 cm of the targeted femoral or sciatic nerve trunk using the approaches described above. Test stimulation (asymmetric charge-balanced biphasic pulse train, 100 Hz) was delivered to confirm that comfortable stimulation-evoked sensations could be evoked in the regions of RLP and/or PLP. Stimulation intensity was adjusted by changing the amplitude (1–30 mA) and/or pulse width (10–200 µs). If no sensations or undesired sensations (eg, local or distal motor activation, local or distal discomfort) were evoked, the test needle was redirected in small increments until comfortable sensations were evoked that covered the regions of pain. Once the location was optimized, the needle electrode was removed and a fine-wire coiled lead (MicroLead, SPR Therapeutics, Cleveland, Ohio) preloaded in a 20G introducer needle was directed to the same location. If correct lead location was confirmed by again evoking comfortable sensations in the regions of RLP and/or PLP, the lead was deployed by withdrawing the needle introducer while applying pressure at the skin surface. The lead was coiled outside the skin, trimmed, and the exit site was covered with a bandage (Tegaderm, 3M, St Paul, Minnesota). Throughout the testing and implantation procedure, active stimulation was only applied to subjects in the PNS therapy group. Placebo control subjects underwent implantation under ultrasound guidance, and mock testing procedures were performed but no active stimulation was applied.
Leads were connected to external, wearable pulse generators (SPRINT, SPR Therapeutics) mounted on the body using an adhesive hydrogel pad that also served as the return electrode (figure 1). Subjects in the PNS group received stimulation that was programmed to evoke comfortable sensations in the regions of RLP and PLP using the same waveform and parameter range as the test stimulation, and subjects were permitted to adjust stimulation intensity within a range set for them by the study staff. Subjects in the placebo group received sham stimulation in which the stimulator user interface was identical to the PNS therapy group, but no stimulation was delivered. All subjects were instructed to use the stimulation continuously. Subjects returned to the clinic weekly during the 8-week therapy period for lead site checks, dressing changes, programming optimization, and assessment for adverse events. After 4 weeks, subjects in the placebo group crossed over and began receiving active stimulation for the remaining 4 weeks of the therapy period. Leads were optionally replaced at the time of crossover if it was determined one or more of the original leads implanted without active stimulation testing did not produce comfortable sensations in the regions of pain or clinically significant pain relief when stimulation was turned on. At the end of the 8-week therapy period (up to 60 days total), investigators removed all leads. All subjects were followed monthly for 10 additional months (12 months from the time of lead implantation).
Outcomes assessments
The primary efficacy outcome evaluated the proportions of subjects in the PNS therapy and placebo control groups that were responders with ≥50% reductions in average pain (substantial pain reductions, based on consensus definitions22) during weeks 1–4 of the therapy period in all areas of RLP and/or PLP that had baseline average daily pain diary scores ≥4. The percent reduction in average pain was determined by comparing the average of daily pain scores from 7-day diaries completed throughout the analysis period with the average of daily pain scores in the 7-day baseline diary. Missing diary entries were replaced with recall average pain data or, if no recall data were available, with the average baseline pain score. If >50% of diary entries were missing, the subject was considered a treatment failure. The primary safety outcome was the occurrence of device-related and procedure-related adverse events assessed at all visits. All lead fractures were recorded, and any resulting physiological sequelae were reported as adverse events. Lead fragments (100 μm diameter wire) were monitored in situ as reported previously.19 23
Additional key outcomes included pain relief during the second half of the treatment period (weeks 5–8), durability of pain relief in follow-up measured by question 5 of the Brief Pain Inventory Short Form (BPI-SF), pain interference measured by question 9 of the BPI-SF, Patient Global Impression of Change (PGIC), BDI-II, and medication usage. All primary and secondary efficacy outcomes were collected by a blinded assessor.
Statistical analyses
All patients who met the eligibility criteria at the time of lead placement were included in a full analysis set for analysis of primary and secondary efficacy endpoints. Exploratory analyses were performed in a per-protocol set defined as randomized subjects who underwent implantation, completed ≥50% of diary entries during the primary endpoint period, continued to meet study eligibility during the primary endpoint period, and for subjects in the PNS therapy group reported ≥50% coverage of the regions of pain with comfortable sensations at implantation and/or at one or more weekly visits during the primary endpoint period.
Responder rates for reductions in pain and pain interference in the follow-up period were evaluated in a long-term analysis population. Subjects were considered treatment failures if they terminated early due to a return of pain. Multiple imputation was performed for missed visits using a regression model in PROC MI (SAS, Cary, North Carolina) that included as covariates age, gender, ethnicity, race, time since amputation, level of amputation (above or below knee), and baseline average residual and phantom pain intensities. Primary safety endpoint analysis was performed on a safety population consisting of all subjects who underwent a study procedure.
The study was funded with a grant from the US Department of Defense, and enrollment was completed with 28 subjects at the close of the grant funding period. Prospective follow-up is ongoing and will continue until 1 year after the final subject was enrolled. Statistical analyses were performed through the 6-month follow-up visit, and data are summarized thereafter with statistical analysis pending completion of follow-up. Proportional and categorical data were compared between groups using a two-sided Fisher’s exact test with α=0.05. Continuous data were compared using a two-sample Wilcoxon test with α=0.05. Secondary outcomes were not adjusted for multiple comparisons. Summary statistics are presented as average (SD). Predefined statistical analyses were performed by an independent biostatistician. Adverse events were adjudicated by an independent medical monitor.
Results
Study population
Subjects were enrolled from March 2015 to March 2018, with 47 consenting subjects assessed for eligibility. Of these, 28 subjects were randomized to undergo lead implantation in the PNS therapy group or the placebo control group (figure 2). Two subjects, both in the PNS therapy group, were excluded from efficacy analyses due to changes in eligibility prior to implantation. The full analysis set included 26 subjects (PNS group n=12, placebo group n=14). Seven subjects (five in the PNS group and two in the placebo group) have completed the 12-month follow-up visit, and eight subjects (three in the PNS group and five in the placebo group) have completed the 6-month visit but have not yet completed the 12-month visit and are continuing through the prospective follow-up period (figure 2). Additional analyses were conducted in a per-protocol set that included 23 subjects (PNS group n=10, placebo group n=13). Of the three subjects excluded from the per-protocol population, two failed to complete ≥50% of daily diary entries, and one failed to achieve ≥50% coverage at implantation or at any time during the therapy period despite reprogramming and lead replacement, indicating that the therapy was unable to be delivered as intended.

Subject flow diagram showing the progression of subjects in the full analysis population. Subjects who completed the 6-month visit but have not yet completed the 12-month visit are noted as “in progress.” PNS, peripheral nerve stimulation.
All subjects had a traumatic lower extremity amputation with an average of 8.7 (SD 7.3) years elapsed since the time of injury and 7.0 (SD 6.6) years since the time of amputation (table 1). A majority of subjects (58%, 15 of 26) had amputations above the knee. Two subjects were bilateral amputees, but each subject qualified for lead implantation (baseline RLP and/or PLP ≥4 at baseline) on only one side. A total of 18 subjects reported qualifying RLP with an average baseline score of 6.4 (SD 1.0) in the PNS group (n=7) and 6.4 (SD 1.3) in the placebo group (n=11), and 24 subjects reported qualifying PLP with an average baseline score of 6.9 (SD 1.7) in the PNS group (n=11) and 6.8 (SD 1.7) in the placebo group (n=13). Subjects previously used or were currently using a wide range of opioid, non-opioid, and other therapies for their postamputation pain, most notably opioid oral medications (92%), non-opioid oral medications (92%), and physical therapy (85%).
Demographic and baseline characteristics
Average residual and phantom limb pain
A significantly greater proportion of subjects receiving PNS therapy (58%, 7 of 12, p=0.037) reported ≥50% pain relief compared with subjects receiving placebo therapy (14%, 2 of 14) during weeks 1–4 of the therapy period (figure 3). The primary endpoint evaluated pain in all areas (RLP and/or PLP) that qualified with pain ≥4 at baseline. Some patients qualified to be evaluated for RLP, PLP, or both. Table 2 shows the average reductions in RLP and PLP separately for the subjects who qualified for analysis of each area of pain, although the study was not powered to analyze RLP or PLP individually. In the first half of the therapy period (weeks 1–4), the average reductions in RLP and PLP in the PNS therapy group were 33% (SD 44) and 50% (SD 24), respectively. Among the seven responders (defined based on the primary endpoint criterion of ≥50% pain relief), the average reductions in RLP and PLP were 73% and 69%, respectively.
Summary of residual and phantom limb pain
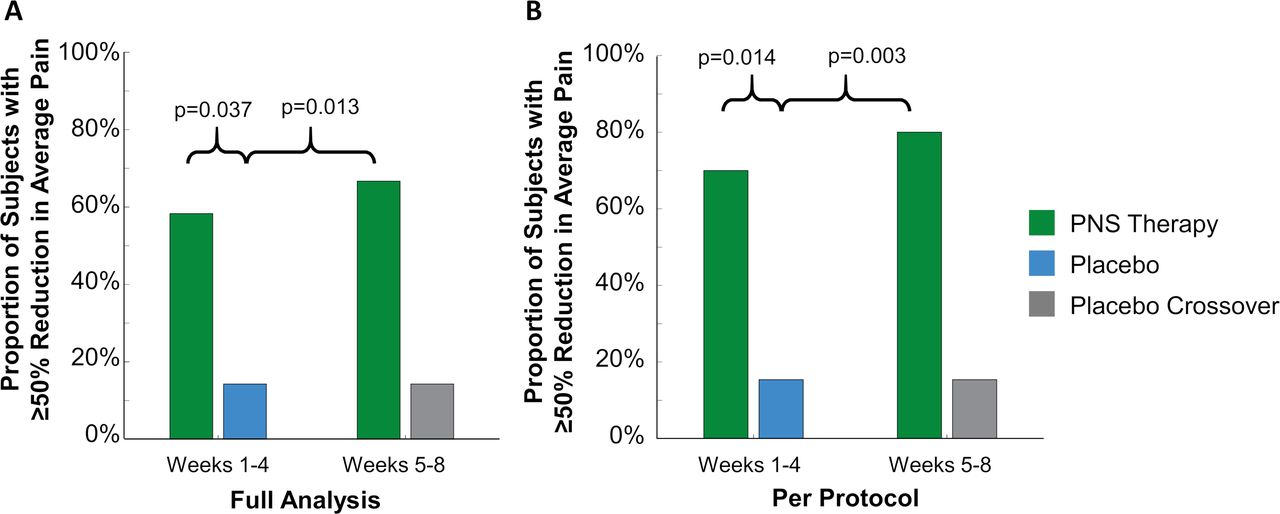
Proportions of subjects with ≥50% reductions in all qualifying regions of residual limb pain and phantom limb pain. Proportions in the (A) full analysis set and (B) per-protocol set during weeks 1–4 and weeks 5–8 of the PNS therapy period were compared with the placebo group at the end of the 4-week placebo period. PNS, peripheral nerve stimulation.
There was also a significant effect of PNS therapy in the per-protocol set, in which 70% (7 of 10, p=0.013) of subjects receiving PNS reported clinically significant pain relief, compared with 15% (2 of 13) of subjects receiving placebo during weeks 1–4. In the first half of the therapy period (weeks 1–4), the average reductions in RLP and PLP among PNS subjects were 48% (SD 43) and 56% (SD 18), respectively.
In the second half of the therapy period (weeks 5–8), the proportion of subjects in the PNS group reporting ≥50% reductions in pain continued to be significantly greater compared with the proportion in the placebo group at the end of the 4-week placebo period in the full analysis set (67% [8 of 12] vs 14% [2 of 14], p=0.014) and per-protocol set (80% [8 of 10] vs 15% [2 of 13], p=0.003) (figure 3). The average pain reductions in RLP and PLP during weeks 5–8 in the PNS therapy group were 36% (SD 32) and 56% (SD 33) in the full analysis set (table 2), and 48% (SD 29) and 62% (SD 28) in the per-protocol set, including an average 56% reduction in RLP and an average 72% reduction in PLP among the seven responders.
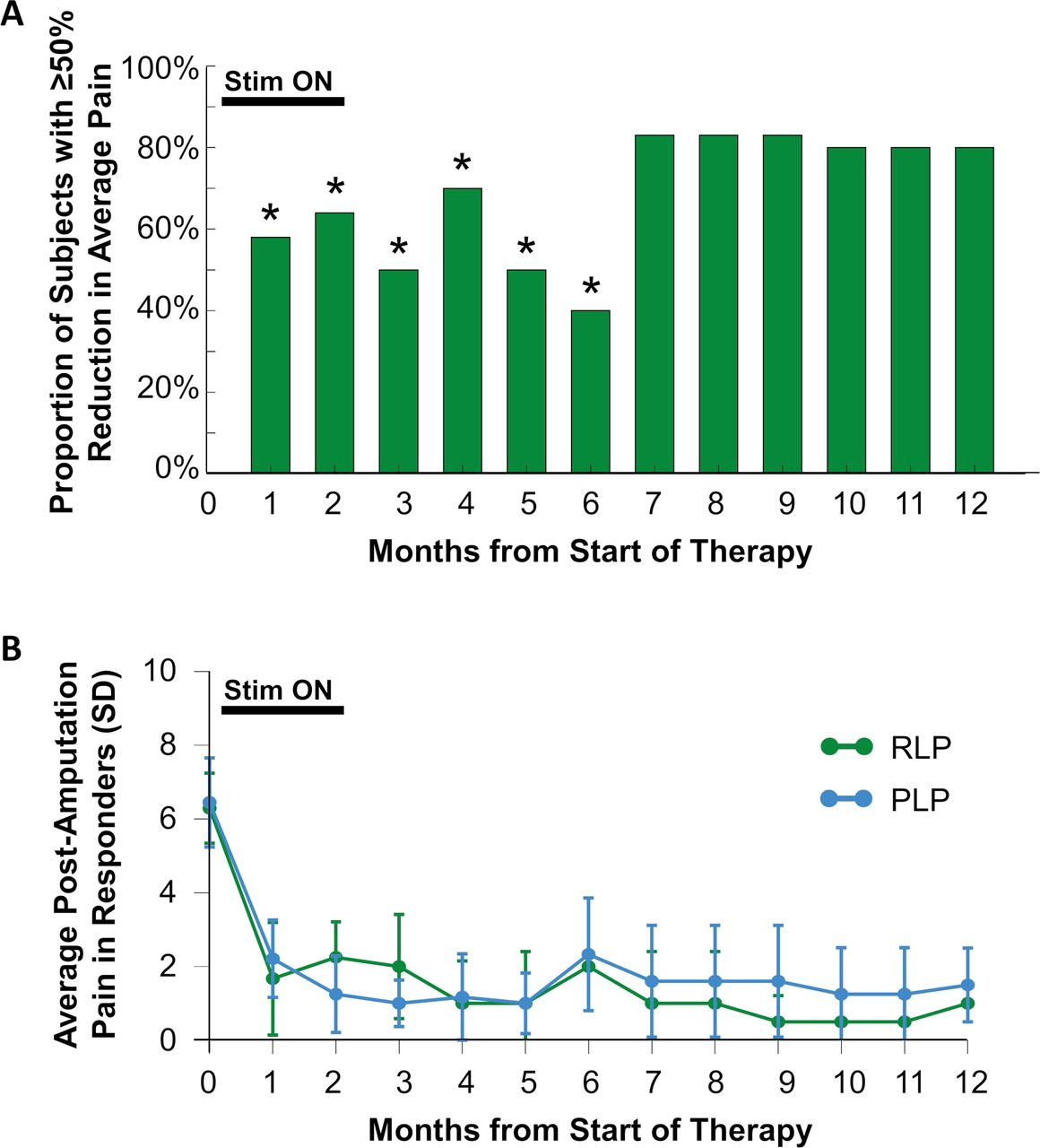
Ten of 12 subjects in the PNS therapy group completed the 8-week therapy period and entered follow-up. Statistically significantly greater proportions of subjects continued to report ≥50% pain relief at months 3–6, compared with the placebo group at the end of the placebo period (figure 4). Prospective follow-up is ongoing at the time of this writing; among subjects who have completed the 12-month follow-up period (10 months after completion of the therapy period and removal of PNS leads), 80% (4 of 5) reported continued substantial pain relief and the average pain reduction in those subjects was 76%.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Durability of pain relief. (A) Proportion of subjects in the peripheral nerve stimulation therapy group reporting ≥50% reduction of average pain at each month. *P<0.05 versus placebo group at the end of the placebo period. (Statistical analysis pending completion of prospective follow-up for months 7–12.) (B) Average residual limb pain (RLP) and phantom limb pain (PLP) scores among responders (those reporting ≥50% reductions in pain at each month of follow-up).
After crossing over at week 4 to begin receiving active stimulation, subjects in the placebo group reported non-significant improvement in average RLP (27% [SD 28] reduction from baseline, p=0.922 compared with weeks 1–4) and statistically significant improvement in average PLP (33% [SD 28] reduction from baseline, p=0.027 compared with weeks 1–4). The proportion of subjects reporting ≥50% reductions in average pain remained 14% (2 of 14).
Twenty-one subjects completed a blinding effectiveness survey. In the PNS therapy group, 8 of 10 (80%) correctly determined that stimulation was active. In the placebo group, 5 of 11 (45%) correctly identified their group assignment, suggesting that the blinding and placebo stimulation methods were effective as a control in this study.
Functional outcomes, global impression of change, and medication usage
Subjects reported functional improvements as measured by reductions in the interference of pain in activities of daily living. A statistically significantly greater proportion of subjects receiving PNS therapy experienced ≥50% reductions in average pain interference in all qualifying regions of RLP and PLP at the end of the treatment period (80%, 8 of 10 p=0.003), compared with the placebo control group at the end of the placebo period (15%, 2 of 13). As noted above, prospective follow-up is ongoing, and a statistically significant majority of subjects in the PNS therapy group continued to report ≥50% reductions in average pain interference at months 3–6 (table 3).
Summary of pain interference, PGIC, and BDI-II
Subjects in the PNS therapy group reported significantly greater PGIC, most notably at the end of the therapy period and the 3-month follow-up (table 3). PGIC was 2.2 (SD 0.9, much improved to very much improved; p=0.007) at the end of the therapy period, compared with 0.6 (SD 1.3, no change to minimally improved) in the placebo group at the end of the placebo period. PGIC in the placebo group increased twofold to 1.3 (SD 1.0, p=0.398) in weeks 5–8 after crossing over to receive PNS. Subjects in the PNS group also reported significantly greater reductions in BDI scores at the end of the therapy period (table 3).
Nine subjects reported opioid use at baseline, including four in the PNS therapy group (average daily morphine equivalent dose [MED]=86.9 [SD 105.2]) and five in the placebo control group (average daily MED=46.8 [SD 70.8]). The average daily MED decreased after 4 weeks (29.2 [SD 24.6] average reduction in MED, p>0.999) and 8 weeks (37.4 [SD 18.0] average reduction in MED, p>0.999) of PNS therapy. These changes were not significantly different from the decrease in average daily MED reported in the placebo group (15.6 [SD 36.7] average reduction in MED).
Among subjects receiving PNS, 33% (3 of 9) stopped use of at least one non-opioid pain medication that was in use at baseline, all three of whom were no longer using any non-opioid pain medications at the end of the therapy period. Among subjects receiving placebo, 25% (3 of 12) also stopped using at least one non-opioid pain medication, but none completely eliminated non-opioid medication use by the end of the placebo period. Overall, 50% (6 of 12) of subjects in the PNS group and 43% (6 of 14) of subjects in the placebo group reduced or stopped opioid and/or non-opioid medication use.
Study-related adverse events and technical difficulties
There were no serious or unanticipated study-related adverse events. A total of 22 study-related events were reported in 46% (13 of 28) of subjects who underwent lead implantation. These included skin irritation or redness at the lead exit site (7), adhesive return electrode pad site (3) or bandage site (4), pain due to implantation or stimulation (5), pruritus at the pad site (1), pruritus under the supporting belt (1), and fatigue (1). All study-related adverse events were mild (96%, 21 of 22) or moderate (4%, 1 of 22) in severity and none (0%, 0 of 22) were severe. The one moderate event was pain during stimulation and was resolved by adjusting stimulation parameters. No leads fractured during treatment. In 15% (5 of 34) of leads whose removal was documented, the lead was fractured at the distal tip on removal.19 23 Lead fragments (100 μm diameter wire), which were labeled as magnetic resonance (MR) Conditional following recent testing,24 were observed in situ and no fragment-related sequelae were subsequently reported during follow-up, similar to previous reports.15
Discussion
This study demonstrates that percutaneous PNS may produce substantial, clinically significant pain reductions in individuals with chronic neuropathic pain following amputation. The same subjects reported significant improvements in pain interference and global impression of change. Furthermore, pain reductions and functional improvements were achieved with concurrent reduction of opioid pain medication usage in all PNS therapy group subjects who were using opioids at the start of the study. This is the first evaluation of the effectiveness of a minimally invasive, fully reversible percutaneous PNS system for neuropathic pain following amputation in a randomized, double-blinded, placebo-controlled study.
It has historically been proposed that PNS systems produce pain relief by activating inhibitory mechanisms in the spinal cord.8 9 16 25–27 While PNS is well accepted as an effective treatment for neuropathic pain,8 10 13 25 28 the invasiveness of previous systems and the methods of delivering those systems (eg, complex surgical implantation of leads near or in contact with the target nerve, devices adapted from SCS, risks of lead migration and nerve damage) limited the ability of interventional pain management physicians to treat patients effectively.11–13 It was theorized that a system designed specifically for use in the periphery and non-surgical, reversible implantation of leads remote from the target nerve (0.5–3 cm distant) may enable selective and reliable activation of the large-diameter sensory fibers that are commonly the target of neuromodulation as part of the classic gate-control mechanism. Such selective activation could effectively engage sources of inhibitory control in the spinal dorsal horn and suppress painful signals from the periphery,27 while also reducing or avoiding activation of off-target fibers like small-diameter pain-competent afferents that could hamper the putative pain-relieving mechanism. This study reported a responder rate of 67% and average pain relief of up to 62% overall (up to 72% in responders) at the end of an 8-week therapy, with corresponding improvements in pain interference and patients’ global impressions of improvement. These results suggest that a combination of the design of PNS-specific leads and development of methods enabling remote location of leads may be key factors in providing consistent pain relief in a majority of patients with refractory chronic neuropathic pain.
The improvements in function observed in this study, as measured by the interference of pain on activities of daily living, PGIC, and depression, were particularly notable because of their potential impact on the quality of life and daily function of patients. Patients who received 8 weeks of PNS in the PNS therapy group reported average reductions in pain interference due to RLP and PLP of 4 or more points on a 0–10 rating scale at the end of the therapy period, which more than quadruples the minimal clinically significant threshold of a 1-point change.22 Similarly, the average PGIC reported at the end of the 8-week therapy was in a range proposed to be of important to substantial clinical significance.22 Because disability and quality of life are significant issues for amputees,4 these data suggest that percutaneous PNS can provide additional benefits beyond pain relief by enabling greater function and return to activities of daily living that may have previously been inhibited by chronic neuropathic pain.
Previous studies of PNS for postamputation or post-trauma neuropathic pain have reported long-term relief of pain, but long-term relief was typically associated with permanently implanted systems that were to be used continuously for many years, and replaced or revised as necessary.8 9 13 25 In the present study, a majority of subjects from whom data have been collected at each monthly follow-up (prospective follow-up is ongoing) have continued to report substantial, clinically significant pain relief after the end of the 8-week PNS therapy. By using PNS leads designed to be removed after an 8-week therapy period, this study demonstrates the potential of percutaneous PNS to produce pain relief that endures beyond 12 months in a large subset of subjects with chronic neuropathic pain following amputation.
Recent findings from non-PNS pain therapies appear to contribute to a mechanistic framework by which a temporary percutaneous PNS therapy may produce long-term pain relief following lead removal. Neuropathic pain after amputation has long been associated with maladaptive CNS plasticity,29 but review of recent results following nerve blocks suggests that aberrant plasticity can be transiently reversed by modulating painful signals coming from nerve targets in the periphery.30 Analysis of studies using cutaneous electromyography to provide sensory feedback to the CNS further suggests that providing non-nociceptive input to the CNS can promote beneficial functional plasticity to combat PLP.31 32 It is hypothesized that the percutaneous PNS therapy combines these dual goals, first by modulating painful signals from the periphery directly and/or at the level of the gating mechanism in the spinal cord to reduce pain and enable reversal of maladaptive cortical changes. Then, over the course of 8 weeks, the therapy enables further beneficial CNS plasticity by generating non-nociceptive sensory input to the cortex. The input is perceived as originating in the area of pain because of selective activation of large-diameter fibers in the peripheral nerve that innervate the painful region(s). The PNS therapy may thereby alter the chronic pain state to prolong or prevent the return of pain even after the removal of the stimulating leads. Furthermore, residual and phantom limb pain are both generally considered to be of neuropathic origin,2 3 and other neuropathic pain states have similar features of maladaptive CNS plasticity.29 As such, this putative mechanism could result in percutaneous PNS producing long-term pain relief in other post-traumatic, postsurgical, idiopathic, or pathologic neuropathic pain conditions.20 21
The study had several limitations. The partial crossover design prevented evaluation of placebo effects beyond 4 weeks. However, placebo effects tend to be maximized during an intervention period and diminish in follow-up,33 so the placebo group at week 4 provided a conservative estimate of the maximal placebo effect for comparison with the PNS group follow-up period. Second, while the pain relief and pain interference outcomes were clinically and statistically significant, the sample sizes made some other outcomes difficult to interpret. For example, only 9 of the 26 enrolled subjects were using opioids at baseline, and baseline opioid usage varied greatly. As a result, there was no significant difference in opioid usage reductions between groups, even though the PNS therapy group had greater absolute and percent reductions in average opioid usage. Of note, two subjects in the placebo group had sporadic, low opioid usage at baseline (eg, did not use every day and used <4 MED/day on average) such that small variations in reported doses resulted in large percent changes. The remaining seven subjects had moderate to high (19–244 MED/day) usage and reported continuous usage every day at baseline. Among subjects who used opioids every day at baseline, the average reduction in opioid usage in the placebo group was 0.8%, compared with 48% in the PNS therapy group after 4 weeks of therapy and 71% after 8 weeks.
The proportion of placebo control subjects reporting substantial pain relief did not increase after crossing over to receive stimulation, although the reduction in phantom pain during weeks 5–8 while PNS was being delivered was statistically significantly (p=0.027) greater than the reduction in pain during the placebo period from weeks 1–4 (table 2). Additionally, patient-reported impressions of improvement doubled after crossover (table 3), suggesting that stimulation did positively impact placebo subjects after crossover. Leads were originally implanted without stimulation feedback in placebo control subjects in order to maintain blinding during the placebo period. These original leads were tested with active stimulation at the time of crossover and lead replacement was considered if coverage or comfort were suboptimal, but lead replacement was optional and was not uniformly applied (only 3 of 14 placebo group subjects had both leads replaced at crossover). This difference in the administration of PNS in the placebo group after crossover compared with the group that received PNS from the outset could explain the absence of an increase in the proportion of patients with ≥50% pain relief after crossover. Mandatory lead replacement at the time of crossover may mitigate this limitation in future studies with similar designs. Because patients were blinded even after crossover, it is also possible that their expectations were influenced by receiving minimal pain relief during the placebo period such that the effects of active stimulation were diminished, similar to a nocebo effect. Additional research is required to evaluate the potential nocebo phenomenon.
Lastly, some subjects reported pain relief ≥50% at the final follow-up visit, so pain relief may have lasted longer than the 12-month observation period in this study. Future study designs may consider the length of the follow-up period to observe the potential for ongoing pain relief following this therapy that lasts longer than 12 months.
Conclusions
This multicenter, randomized, double-blinded, placebo-controlled, partial crossover study demonstrates that percutaneous PNS may provide clinically significant pain relief and functional improvements in patients with neuropathic pain following amputation. While prospective follow-up is ongoing, currently available data suggest that durable pain relief is possible following the conclusion of the short-term PNS therapy. These results, coupled with the strong safety profile both in this study and reported elsewhere,17 18 20 21 23 suggest that patients with neuropathic pain following amputation may receive significant benefit from this minimally invasive, reversible percutaneous PNS therapy without the invasiveness and accompanying complications, costs, and risks of a permanently implanted system.
Acknowledgments
The authors wish to thank Jamie Southern Hizer, Brooke Rierson, Baharin Abdullah, Taylor Coffey, and Mandy Mead for their efforts in conducting the study, and Rosemary Zang, Jeannine Ramsey, Lauren Easley, Haley Arnold, Amorn Wongsarnpigoon, Meredith McGee, and Matthew deBock for study support.
References
Footnotes
Presented at Data from this study were presented at the 2018 Napa Pain Conference in Napa, California, in August 16–19, 2018, and at the Military Health System Research Symposium in Kissimmee, Florida, in August 20–23, 2018.
Contributors CG, RR, LK, SC, NC, and JB contributed to the design of the study. CG, BI, JR, SL, MD, CH, AN, and JM collected the data. CG, BI, JR, SL, MD, CH, RR, LK, AN, JM, SC, NC, and JB contributed to drafting and revision of the manuscript.
Funding The study was funded by the Department of Defense (CDMRP/PRORP W81XWH-12-2-0132) and SPR Therapeutics.
Competing interests CG, JR, MD, RR, and LK have consulted for SPR Therapeutics. The institutions of CG, BI, SL, MD, JM, RR, and LK have received funding from SPR Therapeutics for other clinical studies. NC and JB are employees of SPR Therapeutics.
Patient consent for publication Not required.
Ethics approval The study was approved by the FDA under an Investigational Device Exemption and was conducted in accordance with relevant sections of the US Code of Federal Regulations, the Declaration of Helsinki, and the ICH guidelines for good clinical practice. The study protocol and written informed consent were approved by each study site’s institutional review board and the US Army Medical Research and Materiel Command Human Research Protection Office.
Provenance and peer review Not commissioned; externally peer reviewed.