




Abstract
After spinal cord injury, the disruption of blood–spinal cord barrier by activation of matrix metalloprotease is a critical event leading to infiltration of blood cells, inflammatory responses and neuronal cell death, contributing to permanent neurological disability. Recent evidence indicates that fluoxetine, an anti-depressant drug, is shown to have neuroprotective effects in ischaemic brain injury, but the precise mechanism underlying its protective effects is largely unknown. Here, we show that fluoxetine prevented blood–spinal cord barrier disruption via inhibition of matrix metalloprotease activation after spinal cord injury. After a moderate contusion injury at the T9 level of spinal cord with an infinite horizon impactor in the mouse, fluoxetine (10 mg/kg) was injected intraperitoneally and further administered once a day for indicated time points. Fluoxetine treatment significantly inhibited messenger RNA expression of matrix metalloprotease 2, 9 and 12 after spinal cord injury. By zymography and fluorimetric enzyme activity assay, fluoxetine also significantly reduced matrix metalloprotease 2 and matrix metalloprotease 9 activities after injury. In addition, fluoxetine inhibited nuclear factor kappa B-dependent matrix metalloprotease 9 expression in bEnd.3, a brain endothelial cell line, after oxygen–glucose deprivation/reoxygenation. Fluoxetine also attenuated the loss of tight junction molecules such as zona occludens 1 and occludin after injury in vivo as well as in bEnd.3 cultures. By immunofluorescence staining, fluoxetine prevented the breakdown of the tight junction integrity in endothelial cells of blood vessel after injury. Furthermore, fluoxetine inhibited the messenger RNA expression of chemokines such as Groα, MIP1α and 1β, and prevented the infiltration of neutrophils and macrophages, and reduced the expression of inflammatory mediators after injury. Finally, fluoxetine attenuated apoptotic cell death and improved locomotor function after injury. Thus, our results indicate that fluoxetine improved functional recovery in part by inhibiting matrix metalloprotease activation and preventing blood–spinal cord barrier disruption after spinal cord injury. Furthermore, our study suggests that fluoxetine may represent a potential therapeutic agent for preserving blood–brain barrier integrity following ischaemic brain injury and spinal cord injury in humans.
Introduction
The blood–brain barrier including blood–spinal cord barrier is a highly specialized brain endothelial structure of the fully differentiated neurovascular system. The blood–brain barrier is primarily formed by brain endothelial cells, which form a tight seal due to the presence of well-developed tight junction limiting the entry of plasma components and blood cells into the brain or spinal cord. When damaged by various causes including traumatic spinal cord injury, the blood–brain barrier or blood–spinal cord barrier disruption generates neurotoxic products that can compromise synaptic and neuronal functions (Hawkins and Davis, 2005; Abbott et al., 2006; Zlokovic, 2008) and induces the ‘programmed death’ of neurons and glia, leading to permanent neurological deficits (Xu et al., 2001; Noble et al., 2002; Gerzanich et al., 2009). Thus, preventing the blood–spinal cord barrier disruption should be considered as a potential approach for therapeutic interventions after spinal cord injury.
Matrix metalloproteases (MMPs), a family of zinc endopeptidases, are known to degrade extracellular matrix and other extracellular proteins (Sternlicht et al., 1999; Sternlicht and Werb, 2001) and are essential for remodelling of the extracellular matrix, tissue morphogenesis and wound healing (Werb, 1997). However, excessive proteolytic activity of MMP can be detrimental, leading to numerous pathological conditions including blood–brain barrier or blood–spinal cord barrier disruption after ischaemic brain injury and spinal cord injury (Rosenberg et al., 1994, 1995, 1998; Rosenberg and Navratil, 1997; Asahi et al., 2001; Xu et al., 2001; Noble et al., 2002) and inflammation (Mun-Bryce and Rosenberg, 1998a). For example, MMP9 induces proteolytic degradation of blood–brain barrier and white matter components leading to an increase in infarct volume after transient cerebral ischaemia (Asahi et al., 2001). MMP9 also plays a key role in abnormal vascular permeability and inflammation early after spinal cord injury, and blocking of MMP9 activity inhibits vascular permeability, thereby improving functional recovery (Noble et al., 2002). In addition, upregulation of MMP12 after spinal cord injury induces an increase of blood–spinal cord barrier permeability followed microglia/macrophage activation and blood cell infiltration, thereby hindering recovery of motor function (Wells et al., 2003). Furthermore, MMPs have been implicated in neurodegenerative disorders such as multiple sclerosis and Alzheimer’s disease (Yong et al., 1998; Hartung and Kieseier, 2000). Thus, we hypothesized that blocking MMP activity soon after spinal cord injury would prevent the blood–spinal cord barrier disruption, attenuate inflammatory response, reduce cell death and thereby improve functional recovery.
Fluoxetine as a selective serotonin reuptake inhibitor is most commonly prescribed as an anti-depressant. It also exerts anti-inflammatory and pain-relieving effects (Bianchi et al., 1995; Abdel Salam, 2004). In addition, fluoxetine is known to alleviate post-stroke depression (Wiart et al., 2000; Gainotti, 2010), helps motor recovery in stroke patients (Dam et al., 1996; Pariente et al., 2001) and facilitates cognition after traumatic brain injury (Horsfield et al., 2002). Recent reports also show that fluoxetine provides neuroprotective effect via its anti-inflammatory effect after middle cerebral artery occlusion (Lim et al., 2009) and by inhibiting microglial activation in ischaemic injury and MPTP-induced Parkinson disease in animal models (Chung et al., 2011). In addition, fluoxetine prevents lipopolysaccharide-induced degeneration of nigral dopaminergic neurons by inhibiting microglial activation followed oxidative stress (Chung et al., 2010). Based on these observations, fluoxetine appears to have neuroprotective effects after ischaemic brain injury, but the mechanism of its action is unknown. Thus, we hypothesized that fluoxetine might exert neuroprotective effects by preserving blood–spinal cord barrier integrity after spinal cord injury. Here, we examined whether fluoxetine would prevent blood–spinal cord barrier disruption, inhibit MMP activity, attenuate cell death and improve functional recovery after injury.
Materials and methods
Spinal cord injury
Adult male C57BL/6 (18–22 g, Samtako) mice were anaesthetized with chloral hydrate (500 mg/kg) and a laminectomy was performed at the T9 level, exposing the cord beneath without disrupting the dura. The spinous processes of T8 and T11 were then clamped to stabilize the spine, and the exposed dorsal surface of the cord was subjected to moderate contusion injury (50 kdyn force per 500 to 600 μm displacement) using an Infinite Horizons impactor (Infinite Horizons Inc.). The incision sites were then closed in layers and a topical antibiotic (Bacitracin) was applied to the incision site. For the sham-operated controls, the animals underwent a T9 laminectomy without contusion injury. Surgical interventions and postoperative animal care were performed in accordance with the Guidelines and Policies for Rodent Survival Surgery provided by the Animal Care Committee of the Kyung Hee University.
Drug treatment
Fluoxetine (Sigma) dissolved in sterile PBS was immediately administered into injured mice via intraperitoneal injection (10 mg/kg) (Begovic et al., 2004; Anjaneyulu and Chopra, 2006; Sounvoravong et al., 2007) after spinal cord injury and then further treated once a day for 2 weeks for behavioural test or for indicated time points for other experiments. PBS was administered for vehicle control. For the sham-operated controls, the animals underwent a T9 laminectomy without contusion injury and received no pharmacological treatment. Significant side effects resulting from fluoxetine treatment such as changes in body weight or an increase in mortality were not observed during our experiments.
Cell culture
A transformed mouse brain endothelial cell line, bEnd.3 cells were purchased from ATCC. Details on bEnd.3 culture are provided in the Supplementary material.
Tissue preparation
Tissue preparation was performed as previously described (Yune et al., 2007) (Supplementary material).
Western blot
Total protein was prepared with a lysis buffer containing 50 mM Tris-HCl pH 8.0, 150 mM NaCl, 1% NP-40, 0.5% deoxycholate, 0.1% SDS, 10 mM Na2P2O7, 10 mM NaF, 1 μg/ml aprotinin, 10 μg/ml leupeptin, 1 mM sodium vanadate and 1 mM PMSF. Tissue homogenates were incubated for 20 min at 4°C, and centrifuged at 25 000g for 30 min at 4°C. Protein extraction of both the nuclear and cytosolic fractions was performed as previously described (Yune et al., 2004). The protein concentration was determined using the BCA™ assay kit (Pierce). Protein samples (10–40 μg) were separated on SDS–PAGE and transferred to nitrocellulose membrane (Millipore). The membranes were blocked in 5% non-fat skimmed milk or 5% bovine serum albumin in Tris-buffered saline solution with Tween (TBST) for 1 h at room temperature followed by incubation with antibodies against cleaved caspase 3 (1:1000, Cell Signalling Technology), occludin (1:1000, Invitrogen), zona occludens-1 (ZO-1, 1:1000, Invitrogen), nuclear factor kappa B (NF-κB, 1:500, Santa Cruz Biotechnology), inducible nitric oxide synthase (iNOS, 1:10 000, Transduction Laboratory) and COX2 (1:1000, Cayman Chemicals). The primary antibodies were detected with horseradish peroxidase-conjugated secondary antibodies (Jackson ImmunoResearch). Immunoreactive bands were visualized by chemiluminescence using SuperSignal™ (Thermo Scientific). β-tubulin (1:10 000; Sigma) or Histone 3 (1:1000, Cell Signalling Technology) were used as an internal control. Experiments were repeated three times and the densitometric values of the bands on western blots obtained by AlphaImager® software (Alpha Innotech Corporation) were subjected to statistical analysis. Background in films was subtracted from the optical density measurements.
Gelatin zymography
The activity of MMP2 and 9 at 1 day after injury was examined by gelatin zymography based on a previously described method with minor modifications (Noble et al., 2002) (Supplementary material).
Fluorimetric assay for matrix metalloprotease 9 activity
MMP9 activity was measured using the SensoLyte® 520 MMP-9 Assay Kit according to the manufacturer’s protocol (AnaSpec). Briefly, spinal cords were homogenized with the assay buffer containing 0.1% Triton™X-100, and centrifuged for 15 min at 10 000g at 4°C. The supernatant was collected and stored at −70°C until use. To activate MMP prior to assay, the supernatants were incubated with 4-aminophenylmercuric acetate for 1 h at 37°C. Then, MMP-containing sample was added to a 96-well plate (50 μl/well) and MMP9 substrate (50 μl/well) was added to the sample and control wells. The reagents were mixed by shaking the plate gently for 30 s and incubated at 37°C for 60 min. After incubation, 50 μl of stop solution was added and mixed, and then the fluorescence intensity was measured at an excitation wavelength of 490 nm and an emission wavelength of 520 nm.
Measurement of blood–spinal cord barrier disruption
The integrity of the blood–spinal cord barrier was investigated with Evan's Blue dye extravasation according to previous reports (Chen et al., 2008; Tian et al., 2009), with a few modifications. At 1 day after spinal cord injury, 0.5 ml of 2% Evan's Blue dye (Sigma) solution in saline was administered intraperitoneally. Three hours later, animals were anaesthetized and killed by intracardiac perfusion with saline. The T9 spinal cord segment was removed and homogenized in a 50% trichloroacetic acid solution. After homogenization, samples were centrifuged at 10 000g for 10 min, supernatants were collected and fluorescence was quantified at an excitation wavelength of 620 nm and an emission wavelength of 680 nm. Dye in samples was determined as µg/g of tissue from a standard curve plotted using known amounts of dye (Chen et al., 2008). For qualitative examination of Evan's Blue extravasation, the animals were perfused with PBS and subsequently with 4% formaldehyde, as described above. The spinal cords were sectioned 20-μm thick with a cryostat. The fluorescence of Evan's Blue in spinal tissues was observed with a fluorescence microscope and the relative fluorescence intensity was determined by MetaMorph software (Molecular devices).
Immunohistochemistry
Frozen sections were processed for immunofluorescence staining or double labelling with antibodies against ZO-1 (1:300, Invitrogen), CD31 (1:200, Invitrogen), MMP9 (1:100, Santa Cruz Biotechnology), neuron specific enolase (1:100, Dako Corporation), Gr-1 (1:100, Invitrogen), laminin α-1 (1:100, Santa Cruz Biotechnology), or laminin α-4 (1:100, Santa Cruz Biotechnology). FITC or cy3-conjugated secondary antibodies were used (Jackson ImmunoResearch). Nuclei were labelled with DAPI (4′6-diamidino-2-phenylindole) according to the manufacturer’s protocol (Molecular Probes). The immunohistochemistry control study was performed by omission of the primary antibodies; by replacement of the primary antibodies with non-immune, control antibody, and by pre-absorption with an excess (10 µg/ml) of the respective antigens. Some serial sections were also stained for histological analysis with Cresyl violet acetate.
Terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate-biotin nick end labelling staining
One day after injury, serial spinal cord sections (10 μm thickness) were collected every 100 µm and processed for terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate-biotin nick end labelling (TUNEL) staining using an ApopTag® in situ kit (Chemicon International). Diaminobenzidine (DAB) substrate kit (Vector Laboratories) was used for peroxidase staining, and the sections were then counterstained with methyl green. Control sections were incubated in the absence of terminal deoxynucleotidyl transferase enzyme. Investigators who were blind as to the experimental conditions carried out all TUNEL analyses. TUNEL-positive cells in the grey matter at 1 day after spinal cord injury (total 40 sections) were counted and quantified using a 20× objective. Only those cells showing morphological features of nuclear condensation and/or compartmentalization in the grey matter were counted as TUNEL-positive.
RNA isolation and reverse transcriptase polymerase chain reaction
Total RNA was isolated using TRIzol® Reagent (Invitrogen). Complementary DNA synthesis and reverse transcriptase PCR were performed as previously described (Yune et al., 2007) (Supplementary material).
Enzyme-linked immunosorbent assay
The levels of cytokines (TNFα, IL1β and IL6) were assayed using cytokine ELISA kits (BioSource Europe). Mice were deeply anaesthetized with chloral hydrate (500 mg/kg, intraperitoneally) and cardinally perfused. The spinal cord was immediately removed after perfusion, weighed and frozen in liquid nitrogen for storage at −80°C. To determine the plasma levels of cytokines, blood samples were collected from the femoral vein at 1 day and 5 days after injury, and serum was isolated by centrifugation and stored at −80°C. Tissues were also homogenized and the levels of TNFα, IL1β and IL6 were assayed by ELISA and determined according to the manufacturer’s instructions. All samples were analysed in triplicate.
Fluorescence-activated cell sorting analysis
Flow cytometry was performed as previously described (Stirling and Yong, 2008). Spinal cords (5 mm, one spinal cord per sample) in PBS were mechanically disrupted with a small glass Dounce homogenizer, and single-cell suspensions were obtained by passing the solution through a wire mesh screen (70 µm in size; Sigma). Spinal cord samples were subjected to centrifugation at 4°C at 200g for 10 min (low brake). Pellets were resuspended in foetal bovine serum staining buffer (BD Biosciences) and were subjected to centrifugation (1200g) for 7 min, slow brake at 4°C). Pellets were then resuspended in foetal bovine serum staining buffer. Spinal cord samples were split into several tubes, and cells alone and isotype-matched control samples were generated from 10 μl of each sample (mix of all spinal cord samples) to control for non-specific binding and autofluorescence. The following isotype control antibodies were used: phycoerythrin-labelled rat IgG2b, K, fluorescein isothiocyanate (FITC)-labelled rat IgG2b, K, and peridinin chlorophyll a protein (PerCP)-conjugated rat IgG2b, K (Pharmingen). Cell counts were performed by adding 10 μl of trypan blue to 10 μl of each sample to optimize antibody dilutions. Fc block (BD Biosciences) was added to each sample (10 min at 4°C) to minimize background staining. After incubation with combinations of antibodies for 30 min at 4°C, the samples were washed twice in foetal bovine serum staining buffer and resuspended in 1% buffered formalin. The antibodies used were anti-Gr-1 FITC, anti-CD11b FITC and anti-CD45 PerCP (BD Bioscience). All samples were then immediately analysed with a Becton Dickinson LSR Benchtop Flow Cytometer (BD Biosciences). Forward scatter was adjusted to minimize cellular debris, and propidium iodide exclusion was used to determine cell viability. A minimum of 250 000 cells from spinal cord samples and 50 000 cells from blood samples were analysed.
Behavioural tests
Locomotor outcome after spinal cord contusion injury was assessed using the Basso Mouse Scale (Basso et al., 2006). Mice were scored in an open-field environment by trained investigators who were blind to the experimental conditions. Consensus scores for each animal were averaged at each time point for a maximum of nine points for the Basso Mouse Scale score and 11 points for the subscore, which assesses finer aspects of locomotion.
Statistical analysis
Data, except behaviour tests, are presented as the mean ± SD values and behavioural data presented as the mean ± SEM. Comparisons between vehicle and fluoxetine-treated groups were made by unpaired Student’s t-test. Behavioural scores from the Basso Mouse Scale analysis were analysed by repeated measures ANOVA (Time versus Treatment). Tukey’s multiple comparison was used as a post hoc analysis. Statistical significance was accepted with P < 0.05. Statistical analyses were performed using SPSS 15.0 (SPSS Science).
Results
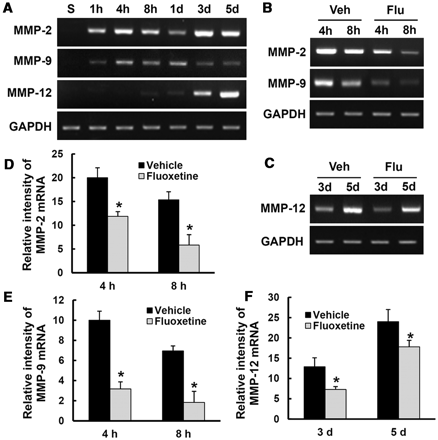
Fluoxetine inhibits the expression and activation of matrix metalloproteinases 2, 9 and 12 after spinal cord injury
Mice were subjected to contusive injury (spinal cord injury) at T9 level and killed at indicated time points after injury. Total RNA and tissue extracts from spinal cord (8 mm in length) including lesion epicentre were prepared as described above. First, we examined MMP2, 9 and 12 messenger RNA expression after injury by reverse transcriptase PCR (n = 3). As shown in Fig. 1A, messenger RNA levels of MMP2, 9 and 12 increased after injury. In addition, fluoxetine significantly inhibited MMP2 and MMP9 messenger RNA expression at 4 and 8 h, and MMP12 at 3 and 5 days after injury as compared to vehicle controls (Fig. 1B–F). Next, we analysed MMP2 and 9 activities by gelatin zymography. As shown in Fig. 2A, MMP9 activity was markedly increased at 8 h and 1 day after injury, with bands corresponding to the MMP9 active form and the inactive zymogen (pro-MMP9). This activity decreased by 5 days after injury. Pro-MM2 appeared in all injured samples and active MMP2 was detected at 5 days after injury (Fig. 2A) as reported (Noble et al., 2002; Hsu et al., 2006; Choi et al., 2010). Furthermore, fluoxetine significantly decreased the level of pro-MMP9, active MMP9 and pro = MMP2 as compared to vehicle controls at 1 day after injury (Fig. 2B and C) (n = 4, pro-MMP9, vehicle, 2.5 ± 0.3 versus fluoxetine, 1.2 ± 0.6; active MMP9, vehicle, 6.8 ± 0.2 versus fluoxetine, 3.0 ± 0.3; pro-MMP2, vehicle, 3.2 ± 0.44 versus fluoxetine, 1.9 ± 0.3, P < 0.05). In addition, we confirmed the effect of fluoxetine on MMP2 and 9 activities by using the fluorimetric enzyme activity assay kit. As shown in Fig. 2D, fluoxetine significantly inhibited MMP9 activity at 1 day after injury as compared to vehicle controls. However, we were unable to show MMP2 activity at 1 day after injury due to its low activity (data not shown). Double labelling shows that MMP9 was localized in neuron specific enolase-positive motor neurons of ventral horn, Gr-1-positive neutrophils and CD31-positive endothelial cells at 1 day after injury (Fig. 2E). However, MMP9 was not detected in uninjured, normal or sham spinal cord (data not shown).
Fluoxetine inhibits disruption of tight junction after spinal cord injury. Total spinal extracts were prepared at the time points indicated (8 h, 1 day, 3 days or 7 days after injury) as described in the Materials and methods. Mice were treated with fluoxetine (10 mg/kg) and total spinal extracts or tissue sections at 1 day after injury were prepared (n = 4/group). (A) Western blots of occludin and ZO-1 at 8 h to 7 days after SCI. (B) Western blots of occludin and ZO-1 of vehicle or fluoxetine treated animals at 1 day after injury. (C) Densitometric analyses of western blots. Data represent mean ±SD. *P < 0.05 versus vehicle control. (D) Representative micrographs showing double immunofluorescence with ZO-1 and CD31 antibodies (endothelial cell marker) at 500 mm caudal to the lesion epicentre. Scale bar = 10 μm.
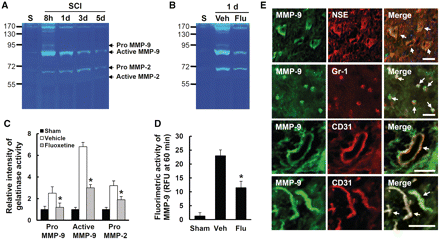
Fluoxetine inhibits MMP2 and 9 activation after spinal cord injury. After spinal cord injury, mice were treated with fluoxetine and gelatin zymography was performed with protein lysates prepared at indicated time points (n = 4/group). (A) Gelatin zymography showing temporal profile of MMP2 and 9 activities after injury. (B) The effect of fluoxetine on MMP2 and 9 at 1 day after injury. (C) Densitometric analyses of zymography. (D) MMP9 activity was measured fluorometrically using a 5-carboxyfluorescein/QXL520 fluorescence resonance energy transfer peptide. The change in fluorescence for 60 min was measured using a luminescence spectrometer (n = 5/group) and indicated as relative fluorescence units (RFU). (E) Representative fluorescence microscopic photographs showing that neuron specific enolase-positive neurons, Gr-1-positive neutrophils and CD31-positive endothelial cells are positive for MMP9 at 1 day after injury. Scale bar = 30 μm. All data represent mean ± SD. *P < 0.05 versus vehicle control. NSE = neuron specific enolase.
Fluoxetine inhibits nuclear factor κB dependent matrix metalloproteinase 9 expression and loss of tight junction proteins in endothelial cells after oxygen–glucose deprivation/reoxygenation
NFκB signalling pathway has been known as one of the MMP9 expression regulators (Tahanian et al., 2011). To investigate how fluoxetine inhibits MMP9 expression in endothelial cells, we applied oxygen–glucose deprivation and reoxygenation to bEnd.3 cells, a mouse brain endothelial cell line. As shown in Fig. 3A, western blots of nuclear extracts with anti-p65 antibody showed that the level of translocated NFκB into the nucleus was markedly increased by 1 h of reoxygenation after oxygen–glucose deprivation, whereas fluoxetine significantly inhibited NFκB translocation (Fig. 3A and B). To confirm the involvement of NFκB, we examined the effect of MG-132 (a proteasome inhibitor that inhibits degradation of IκB and suppresses NFκB translocation to the nucleus) on MMP9 induction. In bEnd.3 cells, MMP9 messenger RNA expression was increased at 1 h reoxygenation after oxygen–glucose deprivation for 6 h as compared with controls, which was reversed by pretreatment with MG-132. In addition, fluoxetine treatment significantly inhibited MMP9 messenger RNA expression (Fig. 3C). These findings indicate that the induction of MMP9 by oxygen–glucose deprivation/reoxygenation occurs through NFκB activation and fluoxetine inhibits MMP9 expression in part by inhibiting the NFκB pathway. Next, we examined whether fluoxetine affects the alteration of tight junction proteins after oxygen–glucose deprivation/reoxygenation injury since blood–brain barrier disruption is known to be associated with degradation of endothelial tight junction proteins as well as decrease in their expression after CNS injuries, including spinal cord injury (Benton et al., 2008; Liu et al., 2009). As shown in Fig. 3D and E, the levels of occludin and ZO-1 decreased at 6 h after reoxygenation. However, the decrease in occludin or ZO-1 level after oxygen–glucose deprivation/reoxygenation was significantly reversed by fluoxetine treatment. Immunocytochemistry also revealed that the intensity of ZO-1 expression decreased by oxygen–glucose deprivation/reoxygenation as compared to oxygen–glucose deprivation/reoxygenation-treated control cells, and fluoxetine treatment prevented the loss of ZO-1 expression (Fig. 3F).
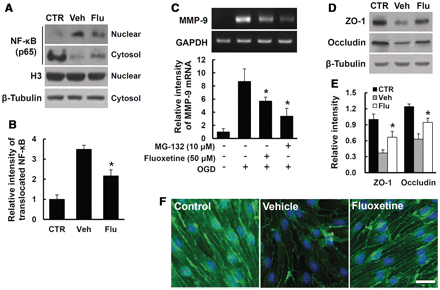
Fluoxetine inhibits NFκB dependent MMP9 expression and loss of tight junction proteins in endothelial cells after oxygen–glucose deprivation/reoxygenation. bEnd.3 cells were pretreated with vehicle, fluoxetine (50 μm) or MG-132 (10 μm) for 30 min before oxygen–glucose deprivation. (A) Western blot analysis of NFκB in nuclear and cytoplasmic extracts using NFκB p65 antibody at 1 h reoxygenation after oxygen–glucose deprivation for 6 h. Beta-tubulin and histone 3 were used as internal controls (CTR) of protein loading for cytosol and nuclear fraction, respectively. (B) Densitometric analyses of western blots for translocated (activated) NFκB. (C) Reverse transcriptase PCR for MMP9 at 1 h reoxygenation after oxygen–glucose deprivation (OGD) for 6 h. (D) Western blots for ZO-1 and occludin in bEnd.3 cell lysate treated with vehicle or fluoxetine (Flu) at 6 h reoxygenation after oxygen–glucose deprivation for 6 h. (E) Densitometric analyses of western blots. (F) Immunocytochemistry from fixed bEnd.3 cells at 6 h reoxygenation after oxygen–glucose deprivation for 6 h. Scale bar = 30 μm. All data represent mean ± SD from five separate experiments. *P < 0.05 versus vehicle control.
Fluoxetine inhibits tight junction disruption after spinal cord injury
Next, we examined the alterations of spinal cord injury-induced tight junction proteins and the effect of fluoxetine on these alterations by western blot. We previously found that the antibodies against occludin and ZO-1 showed specific immunoreactivity at expected molecular weight proteins (65 kDa for occludin, 220 kDa for ZO-1) (data not shown). In addition, the level of occludin or ZO-1 was decreased and the decrease was especially prominent at 1 and 3 days after injury (Fig. 4A). Furthermore, fluoxetine significantly attenuated the decrease in occludin and ZO-1 levels at 1 day after injury as compared with vehicle controls (Fig. 4B and C) (n = 4, occludin, vehicle, 0.27 ± 0.12 versus fluoxetine, 0.83 ± 0.07; ZO-1, vehicle, 0.15 ± 0.08 versus fluoxetine, 0.65 ± 0.05; P < 0.05). Double labelling immunofluorescence also showed that the fluorescence intensity of ZO-1 and CD31 immunoreactivity was decreased after injury as compared to sham controls, and fluoxetine treatment attenuated the decrease in its intensity (Fig. 4D). These data indicate that fluoxetine preserves tight junction integrity by inhibiting degradation of tight junction molecules, and thereby prevents blood–spinal cord barrier disruption after spinal cord injury.
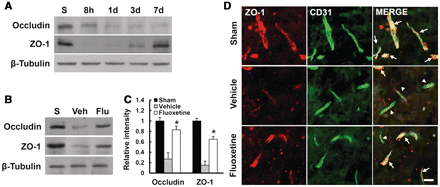
Fluoxetine inhibits disruption of tight junction after spinal cord injury. After injury, mice were treated with fluoxetine (10 mg/kg) and total spinal extracts or tissue sections at 1 day after injury were prepared as described (n = 4/group). (B) Western blots of occludin and ZO-1 at 1 d after injury. (C) Densitometric analyses of western blots. Data represent mean ± SD. *P < 0.05 versus vehicle control. (D) Representative micrographs showing double immunofluorescence with ZO-1 and CD31 (endothelial cell marker) at 500 μm caudal to the lesion epicentre. Scale bar = 10 μm.
Fluoxetine inhibits the increase of blood–spinal cord barrier permeability after spinal cord injury
It is well known that the tight junction in the endothelial cells of blood vessels is involved in the integrity of blood–brain barrier or blood–spinal cord barrier (Zlokovic, 2008). After spinal cord injury, the disruption of blood–spinal cord barrier by MMP activation is also well documented (Noble and Wrathall, 1989). Since fluoxetine inhibited MMP expression and activities (Figs 1 and 2) and preserved ZO-1 and occludin in endothelial cells (Fig. 3), we expected that fluoxetine would inhibit blood–spinal cord barrier permeability after spinal cord injury. Thus, we examined the effect of fluoxetine on blood–spinal cord barrier permeability at 1 day after injury by Evan's Blue assay (n = 5). As shown in Fig. 5A, spinal cord injury caused a marked increase in the amount of Evan's Blue dye extravasation compared with the uninjured, sham controls, which implies blood–spinal cord barrier leakage. Furthermore, fluoxetine (10 mg/kg) treatment significantly reduced the amount of Evan's Blue dye extravasation at 1 day after injury when compared with vehicle controls (n = 5, vehicle, 27.5 ± 0.8 versus fluoxetine, 10.5 ± 1.8, P < 0.05) (Fig. 5B). Qualitative analysis also shows that the fluorescence intensity of Evan's Blue in the injured spinal cord (at 1 day) was higher than sham controls, and fluoxetine significantly reduced the fluorescence intensity (n = 4, vehicle, 118 ± 8.1 versus fluoxetine, 42 ± 3.0, P < 0.05) (Fig. 5C and D). These data indicate that fluoxetine inhibits the blood–spinal cord barrier permeability after injury.
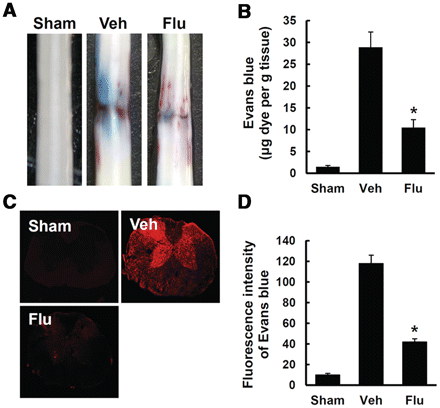
Fluoxetine inhibits the increase in blood–spinal cord barrier permeability after spinal cord injury. After spinal cord injury, mice were treated with fluoxetine and blood–spinal cord barrier permeabitliy was measured at 1 day after injury by using Evan's Blue dye (n = 5/group). (A) Representative whole spinal cords showing Evan's Blue dye permeabilized into spinal cord at 1 day. (B) Quantification of the amount of Evan's Blue. (C) Representative confocal images of an Evan's Blue extravasation at 1 mm caudal to the lesion epicentre at 1 day after spinal cord injury. (D) Quantification of the fluorescence intensity of Evan's Blue. All data represent mean ± SD. *P < 0.05 versus vehicle controls.
Fluoxetine inhibits blood cell infiltration and the expression of cytokines and inflammatory mediators after spinal cord injury
After spinal cord injury, the blood cell infiltration following blood–spinal cord barrier disruption initiates inflammatory responses, leading to the secondary injury cascade by producing inflammatory mediators such as IL1β, IL6, TNFα, COX2 and inducible nitric oxide synthase (Mun-Bryce and Rosenberg, 1998b). Therefore, we examined the effect of fluoxetine treatment on blood cell infiltration by fluorescence-activated cell sorting and expression of inflammatory mediators by reverse transcriptase PCR, western blot, and ELISA assay at indicated time points after injury. In general, neutrophils at 1 day and macrophages at 5 days infiltrated after injury are known to mediate inflammatory responses (Mabon et al., 2000). Thus, we measured the number of blood cells infiltrated into injured spinal cord at 1 and 5 days after injury by fluorescence-activated cell sorting analysis. Only double positive cells (Gr-1/CD45 for neutrophil at 1 day and CD45/CD11b for macrophage at 5 days) were counted and analysed as described (Stirling and Yong, 2008). As shown in Fig. 6, the number of neutrophils and macrophages infiltrated was dramatically increased after injury as compared with sham controls, and fluoxetine significantly attenuated the increase in the blood cell infiltration as compared with vehicle controls at indicated time points after injury (n = 4, CD45/Gr-1-positive cells, vehicle, 100 ± 4.7 versus fluoxetine, 23.8 ± 6.7%; CD45/CD11b-positive cells, vehicle, 99.5 ± 10.7 versus fluoxetine, 32.2 ± 2.9%; P < 0.05). In addition, the expression levels of TNFα, IL1β (at 2 h), IL6, inducible nitric oxide synthase and COX2 (at 6 h) messenger RNA were upregulated after injury, but fluoxetine treatment significantly reduced their levels as compared with vehicle controls (n = 3) (Fig. 7A and B). ELISA assays also showed that fluoxetine significantly inhibited the production of TNFα, IL1β and IL6 1 day after injury (TNFα, IL1β, and IL6 of vehicle group: 39 ± 4.4 pg/ml, 45 ± 2.8 pg/ml, 19 ± 1.8 pg/ml versus fluoxetine group: 22 ± 0.9 pg/ml, 31 ± 2.6 pg/ml, 12 ± 2.1 pg/ml, respectively; n = 5, P < 0.05) (Fig. 7C). By western blot, the protein levels of inducible nitric oxide synthase and COX2 at 1 day after injury were significantly decreased by fluoxetine treatment as compared with vehicle controls (n = 5, P < 0.05) (Fig. 7D and E). In addition, the plasma levels of cytokines were increased after spinal cord injury, whereas fluoxetine did not affect the plasma levels of these cytokines (Supplementary Fig. 1) (1 day: TNFα, IL1β and IL6 of sham group, 1.7 ± 0.3 pg/ml, 0.4 ± 0.4 pg/ml, 3.3 ± 0.99 pg/ml; vehicle group, 2.3 ± 0.8 pg/ml, 1.4 ± 0.56 pg/ml, 17.5 ± 3.3 pg/ml; fluoxetine group, 2.5 ± 2.0 pg/ml, 1.2 ± 0.6 pg/ml, 16.8 ± 2.7 pg/ml, respectively; n = 10, P < 0.05). This suggests that fluoxetine may not affect systemic inflammation responses after injury.
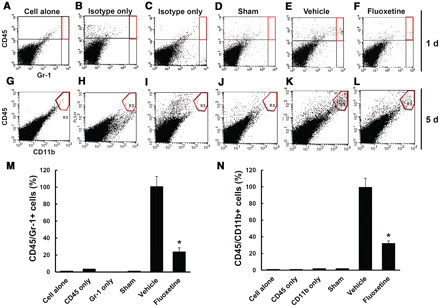
Fluoxetine inhibits infiltration of neutrophils and macrophages after spinal cord injury. After spinal cord injury, mice were treated with fluoxetine (10 mg/kg) and blood cells infiltrated into injured spinal cord at 1 and 5 days were prepared and analysed by flow cytometry after staining with Gr-1/CD45 (neutrophil markers) and CD11b/CD45 (macrophage markers) antibodies (n = 4/group). Density plots of the gated region without fluorescent-conjugated antibodies (A, G) or with isotype-matched control antibodies (B, C, H, I) as control experiments, and density plot of representative sham (D, J), vehicle (E, K) and fluoxetine (F, L) treated spinal cord samples. (M–N) Quantification of Gr-1high/CD45high for neutrophils (M) at 1 day and CD11bhigh/CD45high for macrophages (N) at 5 days after injury as a percentage of vehicle controls. Data represent mean ± SD. *P < 0.05 versus vehicle controls.
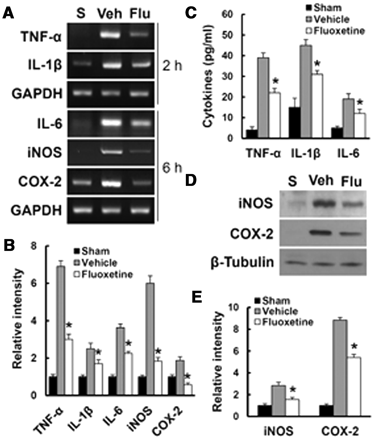
Fluoxetine inhibits the expression of cytokines and inflammatory mediators after spinal cord injury. Total RNA and protein extracts from vehicle or fluoxetine-treated spinal cords at indicated time points after injury were prepared. (A) Reverse transcriptase PCR of TNFα, IL1β (at 2 h), IL6, COX2 and inducible nitric oxide synthase (iNOS) (at 6 h) in sham, vehicle-treated and fluoxetine-treated spinal cords after injury (n = 3/group). (B) Quantitative analysis of reverse transcriptase PCR. (C) ELISA of TNFα, IL1β, IL6 at 1 day in sham, vehicle-treated and fluoxetine-treated spinal cords after injury (n = 5/group). (D) Western blots of inducible nitric oxide synthase and COX2 at 1 day in sham, vehicle-treated and fluoxetine-treated spinal cords after injury (n = 5/group). (E) Quantitative analyses of western blots. Data represent mean ± SD. *P < 0.05 versus vehicle controls.
Fluoxetine inhibits the expression of chemokines induced after spinal cord injury
The expression of chomokines such as Gro-α (CXCL1), MCP1 (CCL2) and MIP1α (CCL3) is known to increase early after spinal cord injury and induces the infiltration of blood cells such as neutrophils and macrophages, thereby facilitating inflammatory responses (McTigue et al., 1998; Ghirnikar et al., 2001; Ousman and David, 2001). Since our data showed that fluoxetine reduced the number of infiltrated blood cells after spinal cord injury, we anticipated that fluoxetine would decrease chemokine expression after spinal cord injury. Thus, we examined the effect of fluoxetine on the expression of chemokines such as Gro-α, MIP2α (CXCL2), MCP1, MIP1α and MIP1β (CCL4) by reverse transcriptase PCR (n = 3). As shown in Fig. 8A, the expression of Gro-α, MIP2α and MCP1 messenger RNA increased at 4 h, 8 h and 1 day after injury, respectively, and then decreased while the expression of MIP1α and MIP1β messenger RNA increased 4 h after injury and then maintained up to 5 days after injury, as reported (McTigue et al., 1998; Ousman and David, 2001). Furthermore, messenger RNA expression of Gro-α, MIP1α and MIP1β at 8 h after injury was significantly inhibited by fluoxetine treatment (Fig. 8B and C, n = 3). However, MIP2α and MCP1 messenger RNA expression was not affected by fluoxetine (data not shown). These data suggest that fluoxetine might reduce the number of infiltrating blood cells by attenuating the expression of chemokines such as Gro-α, MIP1α and MIP1β after spinal cord injury.
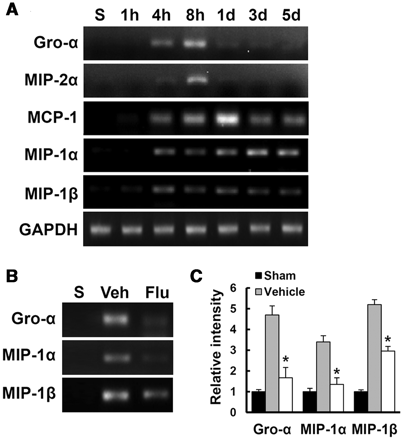
Fluoxetine inhibits the expression of chemokines induced after spinal cord injury. Total RNA from vehicle or fluoxetine-treated samples at indicated time points after injury were prepared (n = 3/group). (A) Reverse transcriptase PCR of Gro-α, MIP2α, MCP1, MIP1α and MIP1β messenger RNA expression after injury. (B) The effect of fluoxetine on Gro-α, MIP1α and MIP1β expression at 1 day after injury. (C) Quantitative analysis of reverse transcriptase PCR. Data represent mean ± SD. *P < 0.05 versus vehicle controls.
Fluoxetine inhibits caspase 3 activation and apoptotic cell death after spinal cord injury
Recent reports show that fluoxetine provides a neuroprotective effect by its anti-inflammatory effect after middle cerebral artery occlusion (Lim et al., 2009) and by inhibiting microglial activation in ischaemic and MPTP-induced Parkinson disease model (Chung et al., 2011). Thus, we examined the effect of fluoxetine on apoptotic cell death in the grey matter at 1 day after spinal cord injury by TUNEL staining. Serial transverse sections (10 μm thickness) were collected every 100 µm from 2 mm rostral to 2 mm caudal to the lesion epicentre (total 40 sections for neurons). TUNEL-positive cells were observed mostly near and within the lesion area in the grey matter at 1 day (Fig. 9A). Fluoxetine treatment significantly decreased the number of TUNEL-positive cells when compared with the vehicle-treated controls (vehicle, 133.5 ± 12.3 versus fluoxetine, 51.5 ± 8.5 cells, n = 5, P < 0.05) (Fig. 9B). In this regard, double-labelling confirms that most TUNEL-positive cells in the grey matter were neurons at 1 day after injury (data not shown) as previously reported (Yune et al., 2009; Lee et al., 2010). In addition, the level of cleaved (activated) forms of caspase 3 increased at 4 h after injury (Lee et al., 2010) and fluoxetine significantly decreased the level of activated caspase 3 at 4 h after injury when compared with vehicle controls (Fig. 9C and D) (vehicle, 4.3 ± 0.4 versus fluoxetine, 2.6 ± 0.25, n = 3, P < 0.05). Thus, our results indicate that fluoxetine inhibits apoptotic cell death after injury.
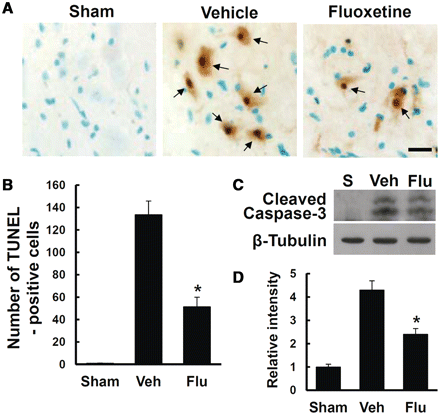
Fluoxetine inhibits caspase 3 activation and apoptotic cell death after spinal cord injury. After spinal cord injury, mice were treated with fluoxetine and spinal tissues and extracts were prepared for TUNEL staining and western blot. (A) Representative images of TUNEL staining at 1 day after spinal cord injury. Scale bar = 20 μm. (B) Quantitative analysis of TUNEL-positive cells (n = 5/group). (C) Western blots of cleaved caspase 3 at 4 h after spinal cord injury. (D) Quantitative analyses of western blots (n = 3/group). Data represent means ± SD. *P < 0.05 versus vehicle controls.
Fluoxetine improves functional recovery after spinal cord injury
After spinal cord injury, mice were immediately treated with fluoxetine (10 mg/kg, intraperitoneally) and further treated once a day for 2 weeks. Functional recovery was then evaluated using the 9-point Basso Mouse Scale score and 11-point Basso Mouse Scale subscore (Basso et al., 2006) for locomotion. As a result, fluoxetine treatment significantly increased the hindlimb locomotor function 10 to 28 days after injury, compared with that observed in vehicle-treated controls (n = 15/group, 28 days, Basso Mouse Scale score, fluoxetine, 7.0 ± 0.26 versus vehicle 4.3 ± 0.31; Basso Mouse Scale subscore, fluoxetine, 4.5 ± 0.3 versus vehicle 0.9 ± 0.22, P < 0.05) (Fig. 10A and B).
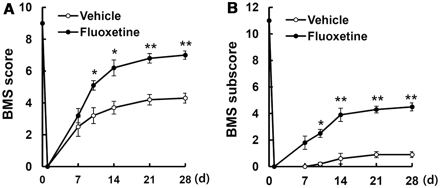
Fluoxetine improves functional recovery after spinal cord injury. After spinal cord injury, mice were treated with fluoxetine and functional recovery was assessed with the Basso Mouse Scale (BMS) score (A) and Basso Mouse Scale subscore (B). Each value represents the mean ± SEM (n = 15/group). *P < 0.05, **P < 0.01 versus vehicle controls.
Discussion
In the present study, we demonstrate that fluoxetine, known as a representative anti-depressant drug, exerts neuroprotective effects by preventing blood–spinal cord barrier disruption and inhibiting MMP activation after spinal cord injury. In addition, we show that fluoxetine reduces the number of infiltrating blood cells, such as neutrophils and macrophages, after injury by inhibiting expression of chemokines such as Gro-α, MIP1α and MIP1β, resulting in reduced inflammatory responses. Furthermore, post-injury treatment with fluoxetine inhibited apoptotic cell death and improved functional recovery after spinal cord injury. Here, we present clear evidence for the mechanism of action of fluoxetine on the expression and/or activity of MMP, which affects the blood–brain barrier/blood–spinal cord barrier integrity after injury. Our findings have important implications in traumatic and ischaemic brain injuries and spinal cord injury in which the disruption of the blood–brain barrier integrity triggers a secondary degenerative cascade including inflammation in pathological processes (Yang et al., 2007; Yu et al., 2008; Higashida et al., 2011; Hosokawa et al., 2011; Miyazaki et al., 2011).
Under physiological conditions, the blood–brain barrier/blood–spinal cord barrier represents a tight barrier between the circulating blood and CNS and is formed by dense tight junction proteins, which seal the space between adjacent brain endothelial cells. Disruption of the blood–brain barrier occurs under various pathological conditions such as stroke and spinal cord injury, leading to an increased cerebrovascular permeability with subsequent development of tissue oedema (Utepbergenov et al., 1998). Although many factors are known to contribute to blood–brain barrier disruption, MMPs play a critical role in the blood–brain barrier/blood–spinal cord barrier disruption in pathological conditions (Asahi et al., 2001; Rosenberg, 2002; Jin et al., 2011). Here, our results show that messenger RNA expression and enzyme activity of MMPs were upregulated after spinal cord injury. Furthermore, fluoxetine treatment significantly inhibited the expression and activity of MMPs after injury (Figs 1 and 2). After spinal cord injury, upregulation of MMP9 has been implicated in spinal cord injury-induced secondary damage and blood–spinal cord barrier disruption by degrading the basal components of blood–brain barrier and facilitating immune cell infiltration (Noble et al., 2002). MMP9 is also shown to mediate hypoxia-induced vascular leakage in the brain via tight junction rearrangement (Bauer et al., 2010). Several reports have also demonstrated that the upregulation of MMP2 contributes to the initial opening of the blood–brain barrier by degrading the basal lamina leading to neuronal injury (Heo et al., 1999; Chang et al., 2003). In addition, minocycline treatment protects the blood–brain barrier and reduces oedema following intracerebral haemorrhage in the rat, by inhibiting MMP12 messenger RNA expression (Wasserman and Schlichter, 2007). MMP12 is also known to be upregulated after spinal cord injury and mechanistically, there is decreased permeability of the blood–spinal cord barrier and reduced neutrophil and macrophage density in MMP12 null mice when compared with wild-type controls (Wells et al., 2003). Thus, these results suggest that fluoxetine effectively prevents blood–spinal cord barrier disruption, in part by inhibiting the expression and activity of MMPs after spinal cord injury.
It is known that tight junction proteins such as occludin, claudin 5 and ZO-1 are essential components of the blood–brain barrier or blood–spinal cord barrier and known to be substrates of MMPs (Yang et al., 2007). Our data show that occludin and ZO-1 were rapidly degraded at soon (8 h to 1 day) after spinal cord injury (Fig. 4A) and fluoxetine significantly inhibited degradation of these molecules (Fig. 4B and C). It has been shown that blood–brain barrier disruption induced by transient focal cerebral ischaemia is attenuated in MMP9 knockout mice by reducing degradation of ZO-1 protein compared with wild-type mice (Asahi et al., 2001). MMP2 and 9 secreted by leukaemic cells also increase the permeability of the blood–brain barrier by disrupting tight junction proteins (Feng et al., 2011). Thus, these results indicate that fluoxetine might prevent blood–spinal cord barrier disruption by reducing degradation of tight junction proteins via inhibition of MMP2 and 9 activities after spinal cord injury. On the other hand, it is not known whether MMP12 could cleave tight junction proteins, although it is shown to cleave elastin, one of the matrix proteins (Fulcher and Van, 2011). However, we can’t rule out the possibility that upregulation of MMP12 may be involved in the degradation or cleavage of tight junction proteins, thereby disruptingthe blood–brain barrier integrity after spinal cord injury.
It is known that the infiltration of blood leukocytes is mediated through two passage processes: the passage of the vascular wall into perivascular spaces and the passage of the glia limitans. Since the respective basement membranes are composed of different laminin isoforms and only the parenchymal basement membrane is linked to dystroglycan (Sixt et al., 2001), the broad MMP inhibitor BB-94 blocks the second step only (Toft-Hansen et al., 2006). Thus, it is very important to define these processes in various vascular diseases, such as autoimmune diseases including multiple sclerosis (Sixt et al., 2001; Agrawal et al., 2006; Wu et al., 2009). However, blood vessels including capillaries are ruptured immediately by mechanical injury itself and further fragmented by various factors such as metalloproteases and SUR1 induced by spinal cord injury (Simard et al., 2007, 2010; Zhang et al., 2011), thereby, further increasing blood infiltration. In this study, we also found that the fragmentation of blood vessels was increased after spinal cord injury as previously reported (Simard et al., 2010). Furthermore, fluoxetine treatment significantly inhibited fragmentation of capillaries as compared with vehicle control (Supplementary Fig. 2).
Chemokines are known to mediate chemotaxis and leukocyte activation (Asensio and Campbell, 1999) and induce blood cells such as neutrophils and macrophages to migrate along concentration gradients to the lesion site (Karpus and Ransohoff, 1998). Extensive evidence has shown that inflammatory cells such as neutrophils and macrophages are infiltrated via blood–spinal cord barrier disruption, increase tissue damage, induce apoptotic cell death and impair functional recovery after injury (Carlson et al., 1998; Beattie et al., 2000; Popovich et al., 2003; Okada et al., 2004). Our data show that chemokines such as Gro-α, MIP1α, MIP1β, MIP2α and MCP1 were upregulated after injury (Fig. 8A) and fluoxetine significantly inhibited upregulation of Gro-α, MIP1α and MIP1β (Fig. 8B and C) and thereby reduced the number of infiltrating blood cells (Fig. 6). In parallel with this result, the expression of inflammatory mediators such as TNFα, IL1β, IL6, inducible nitric oxide synthase, and COX2, was upregulated after injury, which was significantly reduced by fluoxetine (Fig. 7). Thus, these results suggest that fluoxetine may inhibit inflammatory responses by attenuating expression of chemokines and reducing blood cell infiltration following production of inflammatory mediators after spinal cord injury.
Carlson et al. (1998) show that the number of infiltrating neutrophils significantly correlates with the amount of tissue damage after spinal cord injury. Several studies also demonstrate that suppression of neutrophil and macrophage infiltration after injury ameliorates apoptotic cell death and improves functional recovery (Hamada et al., 1996; Taoka et al., 1997; Saiwai et al., 2010). In this regard, our results also show that apoptotic cell death was increased after injury (Fig. 9) and fluoxetine significantly attenuated apoptotic cell death (Fig. 9) and improved functional recovery after spinal cord injury (Fig. 10). Thus, our data suggest that the neuroprotective effect of fluoxetine might be mediated in part by preventing blood–spinal cord barrier disruption following infiltration of neutrophils and macrophages after spinal cord injury.
The neuroprotective effect of fluoxetine can be mediated in part by either its primary effect on immune cells and their activation outside of the brain, or its direct effect on brain cells expressing MMPs, tight junction proteins and/or proinflammatory cytokines, as reported (Pellegrino and Bayer, 2002; Diamond et al., 2006; Roumestan et al., 2007; Fazzino et al., 2009; Li et al., 2011). As shown in Supplementary Fig. 1, fluoxetine does not affect systemic inflammation after injury as indicated by the plasma levels of cytokines. Thus, we postulate that the direct effects of fluoxetine on CNS cells expressing MMPs, tight junction proteins and inflammatory cytokines are likely to be involved in its neuroprotective effect after spinal cord injury, although we can’t exclude the possibility of a primary effect of fluoxetine on immune cells outside of the CNS.
In conclusion, this study examined the protective effect of fluoxetine on MMPs and blood–spinal cord barrier integrity after spinal cord injury. Our study shows that fluoxetine attenuated blood–spinal cord barrier disruption and inhibited MMP2, 9 and 12 activities and improved functional recovery after spinal cord injury. Fluoxetine is currently used as an anti-depressant. Considering the neuroprotective effects of fluoxetine in the animal models of CNS injury and diseases (Dam et al., 1996; Pariente et al., 2001; Horsfield et al., 2002; Lim et al., 2009; Chung et al., 2011), our results suggest that fluoxetine may provide potential therapeutic interventions for preventing blood–brain barrier disruption after ischaemic brain injury and spinal cord injury.
Funding
This study was supported by Brain Research Center of the 21st Century Frontier Research Program (No. 2011K000282, 2011K000291), the Pioneer Research Center Program through the National Research Foundation of Korea (No. 20110001692), and Basic Science Research Program through the National Research Foundation of Korea grant (No. 20110000932) funded by the Ministry of Education, Science and Technology (MEST), the Republic of Korea.
Supplementary material
Supplementary material is available at Brain online.
Acknowledgements
We thank Dr. Hyun-Jong Ahn at the Kyung Hee University for FACS analyses.
Abbreviations
- MMP
matrix metalloprotease
- TUNEL
terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate-biotin nick end labelling


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}