Article Text

Abstract
Background and objectives To describe the safety and tolerability of intravenous meloxicam compared with placebo across all phase II/III clinical trials.
Methods Safety data and opioid use from subjects with moderate to severe postoperative pain who received ≥1 dose of intravenous meloxicam (5–60 mg) or placebo in 1 of 7 studies (4 phase II; 3 phase III) were pooled. Data from intravenous meloxicam 5 mg, 7.5 mg and 15 mg groups were combined (low-dose subset).
Results A total of 1426 adults (86.6% white; mean age: 45.8 years) received ≥1 dose of meloxicam IV; 517 (77.6% white; mean age: 46.7 years) received placebo. The incidence of treatment-emergent adverse events (TEAEs) in intravenous meloxicam and placebo-treated subjects was 47% and 57%, respectively. The most commonly reported TEAEs across treatment groups (intravenous meloxicam 5–15 mg, 30 mg, 60 mg and placebo, respectively) were nausea (4.3%, 20.8%, 5.8% and 25.3%), headache (1.5%, 5.6%, 1.6% and 10.4%), vomiting (2.8%, 4.6%, 1.6% and 7.4%) and dizziness (0%, 3.5%, 1.1% and 4.8%). TEAE incidence was generally similar in subjects aged >65 years with impaired renal function and the general population. Similar rates of cardiovascular events were reported between treatment groups. One death was reported (placebo group; unrelated to study drug). There were 35 serious adverse events (SAEs); intravenous meloxicam 15 mg (n=5), intravenous meloxicam 30 mg (n=15) and placebo (n=15). The SAEs in meloxicam-treated subjects were determined to be unrelated to study medication. Six subjects withdrew due to TEAEs, including three treated with intravenous meloxicam (rash, localized edema and postprocedural pulmonary embolism). In trials where opioid use was monitored, meloxicam reduced postoperative rescue opioid use.
Conclusions Intravenous meloxicam was generally well tolerated in subjects with moderate to severe postoperative pain.
Trial registration numbers NCT01436032, NCT00945763, NCT01084161, NCT02540265, NCT02678286, NCT02675907 and NCT02720692.
- meloxicam
- COX-2 inhibitor
- safety
- postoperative pain
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, an indication of whether changes were made, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Meloxicam, a long-acting preferential cyclooxygenase (COX)-2 inhibitor, possesses analgesic, antipyretic and anti-inflammatory activities.1 2 A nanocrystal formulation of meloxicam that can be administered by intravenous bolus injection was developed for the management of moderate to severe pain. Intravenous meloxicam was evaluated in four phase II and three phase III postoperative studies, comprising subjects undergoing dental impaction surgery,3 open abdominal hysterectomy,4 abdominal laparoscopic surgery,5 bunionectomy,6 7 abdominoplasty8 and major surgery.9 In these studies, intravenous meloxicam demonstrated onset of analgesia within 15 min after administration with maintenance over a 24-hour dosing interval.3 4 6–8 Safety findings from individual studies revealed the incidence of adverse events was comparable with placebo,3–9 and where evaluated, intravenous meloxicam was associated with reduced opioid consumption.3 4 6–9 Intravenous meloxicam is an investigational product that has not been approved by the US Food and Drug Administration.
This pooled analysis was performed to describe the safety and tolerability of intravenous meloxicam compared with placebo across all phase II/III postoperative clinical trials. Considering the pathophysiologic importance of COX in prostanoid biosynthesis and subsequent effects on platelet aggregation, vasoconstriction and gastrointestinal (GI) and renal homeostasis,10 this analysis includes examination of safety concerns associated with COX inhibition, including bleeding, cardiovascular and renal events.
Methods
Study design and subjects
Data from seven postoperative studies3–9 are included in the current analysis (online supplemental digital table 1). All subjects who received ≥1 dose of intravenous meloxicam or placebo are included in the safety set. Data from subjects who received active controls (ibuprofen: n=50 [NCT00945763]3; morphine: n=47 [NCT01084161]4; and ketorolac: n=8 [NCT01436032]5) are not included. Subjects (n=56) who switched from placebo in the double-blind phase to intravenous meloxicam in the open-label phase of study NCT010841614 were counted twice (once in the placebo group and once in the intravenous meloxicam group). For subjects (n=21) randomized to receive morphine in the double-blind phase and then switched to intravenous meloxicam in the open-label phase of that same study, data were generated, while the subjects on intravenous meloxicam are included. In NCT01436032,5 two intravenous meloxicam dose groups (7.5 mg and 15 mg) received study medication twice daily. Subjects on these regimens were grouped by their total daily dose.
Supplemental material
Subject disposition, demographics and baseline clinical characteristics in pooled phase II/III clinical trial program
Evaluations
Safety evaluations included treatment-emergent adverse events (TEAEs), vital signs, concomitant medications, clinical laboratory parameters, 12-lead ECGs, wound site evaluations, physical examination and/or opioid consumption (online supplemental digital table 1). All evaluations were made by individual investigators and were not derived from consensus definitions. Investigators determined TEAE severity and the relationship of TEAEs with the study drug.
In the phase III studies, investigators provided wound-healing satisfaction scores using an 11-point numeric rating scale anchored by ‘completely unsatisfied’ (0) and ‘completely satisfied’ (10). In addition, investigators rated the symptoms and severity of erythema, drainage, edema, induration and hematoma using a 5-point Likert scale. Investigators assessed parameters rated with a score >0 for clinical significance. If individual investigators considered the features under evaluation to be clinically significant, a TEAE was recorded.
Laboratory values and vital signs (heart rate, respiratory rate, blood pressure and body temperature) were collected at various time points during the postoperative studies. Changes in vital signs assessed by the investigator to be clinically meaningful were recorded as TEAEs. Because of the differences in data collection time points, the clinical laboratory evaluation presented herein focuses on the pooled phase III studies.
Statistical methods
No missing data imputation was performed. Data from the intravenous meloxicam 5 mg, 7.5 mg and 15 mg groups were combined (low-dose subset). Descriptive statistics are presented; inferential statistics were not produced. Analyses were performed using SAS for Windows (V.9.3 or higher).
Results
Disposition/demographics
The postoperative population included 517 adults who received placebo and 1426 adults who received intravenous meloxicam (fixed doses, 5–60 mg) for the management of moderate to severe postoperative pain (table 1). Most subjects in the meloxicam groups (1045/1426; 73.3%) received ≥2 doses, including 142/327 (43.4%), 814/910 (89.5%) and 89/189 (47.1%) subjects in the meloxicam 5–15 mg, 30 mg and 60 mg groups, respectively.
Demographics and baseline clinical characteristics are summarized in table 1. Most subjects were white, female and aged<65 years; most had no history of hypertension, diabetes or GI disorders. A minority (<7%) were aged >65 years and had impaired renal function (glomerular filtration rate 60‒89 mL/min/1.73 m2).
Treatment-emergent adverse events
Fifty-seven percent of subjects in the placebo group and 47% of meloxicam-treated subjects experienced ≥1 TEAE (table 2). Across all treatment groups (meloxicam 5–15 mg, 30 mg, 60 mg and placebo, respectively), study investigators assessed TEAEs to be mild (23.2%, 44.4%, 20.6% and 45.5%) or moderate (11.6%, 21.6%, 9.0% and 22.6%) in intensity. The most commonly reported TEAEs were nausea, headache, vomiting and dizziness. Among subjects treated with intravenous meloxicam 30 mg, constipation occurred at a higher rate among subjects >65 years with impaired renal function versus the general population (table 3). Approximately one-third of subjects in the placebo group experienced one or more treatment-related TEAE, as did approximately one-fifth of subjects who received any dose of intravenous meloxicam (table 2).
Treatment-emergent adverse events (TEAEs) and treatment-related* TEAEs occurring in ≥3% in any treatment group in pooled phase II/III clinical trial program
Treatment-emergent adverse events (TEAEs) occurring in ≥3% in any treatment group in pooled phase II/III clinical trial program by risk category*
Deaths, serious adverse events (SAEs) and discontinuations
One death was reported in these studies. A 39-year-old placebo-treated woman died suddenly 55 days after treatment in study NCT02675907.7 Death was attributed to ‘complications of the toxic effects of methamphetamine’ and was considered by the investigator to be unrelated to study drug.
A total of 35 subjects experienced SAEs during the conduct of the clinical studies (placebo: n=15; intravenous meloxicam 15 mg: n=5; intravenous meloxicam 30 mg: n=15). Investigators considered the SAEs experienced by 31 subjects to be unrelated to study drug. There were four events (postprocedural hemorrhage, postprocedural pulmonary embolism, jejunal stenosis and anastomotic ulcer) occurring in three placebo-treated subjects that were assessed as possibly related to study drug.
Six subjects had ≥1 TEAE that led to treatment or study discontinuation: 3/517 (0.6%) subjects in the placebo group (postoperative anemia, drug toxicity, postprocedural hemorrhage and postprocedural pulmonary embolism), 1/327 (0.3%) subjects in the intravenous meloxicam 5–15 mg group (rash) and 2/910 (0.2%) subjects in the intravenous meloxicam 30 mg group (localized edema and postprocedural hemorrhage).
Adverse events of special interest (AEOSI)
NSAIDs such as meloxicam have been associated with an increased risk of cardiovascular, thrombotic and GI AEs (together, AEOSIs).11 Sponsor-defined AEOSIs occurring in >1 subject are summarized in table 4. Additional details for AEOSI that were reported and evaluated by the investigator as unrelated to study drug are included in online supplemental digital appendix 1.
Supplemental material
Adverse events of special interest (AEOSI) occurring in >1 subject in any treatment group in pooled phase II/III clinical trial program
Bleeding events (anemia, hematoma, hemorrhage or ecchymosis) are summarized in table 5. Anemia events were reported for 7/517 (1.4%) subjects who received placebo, 26/327 (8.0%) subjects who received intravenous meloxicam 5–15 mg, 23/910 (2.5%) subjects who received intravenous meloxicam 30 mg and 18/189 (9.5%) subjects who received intravenous meloxicam 60 mg. Five subjects (1 [0.2%] placebo and 4 [0.3%] intravenous meloxicam 30 mg) required blood transfusions. Among intravenous meloxicam-treated subjects, four had hemoglobin and hematocrit values below the normal range prior to surgery that decreased further after surgery. The report of severe anemia, which occurred in a placebo subject, was due to a postprocedural hemorrhage (recorded as an SAE). Most subjects with anemia were either prescribed medications (eg, ferrous sulfate; 33/74 [44.6%]) or received no anemia treatment (35/74 [47.3%]). Anemia was reported in 8/153 (5.2%) subjects with a history of GI conditions (esophageal reflux disease, GI ulcer and/or bleeding within the GI tract) treated with intravenous meloxicam 30 mg. In contrast, anemia was reported in 11/757 (1.5%) subjects without history of GI conditions treated with intravenous meloxicam 30 mg. Among placebo-treated subjects with and without history of GI conditions, anemia was reported in 1/66 (1.5%) and 5/451 (1.1%) subjects, respectively. In the placebo and intravenous meloxicam 30 mg treatment groups, the incidence of anemia was higher among subjects who used antithrombotic medications than among those who did not (table 6). The increase in rates of anemia associated with concomitant use of antithrombotic medications was proportional for subjects in the intravenous meloxicam 30 mg and placebo groups compared with subjects in the intravenous meloxicam 30 mg and placebo groups who did not use these medications.
Treatment-emergent bleeding events occurring in >1 subject in any treatment group in pooled phase II/III clinical trial program
Treatment-emergent adverse events (TEAEs) occurring in ≥3% of subjects in any treatment group overall by history of the use of antithrombotic medication in pooled phase II/III clinical trial program
Across all treatment groups, hematoma events (postprocedural or wound) were reported for 2 (0.6%), 3 (0.3%), 1 (0.5%) and 0 subjects in the in the intravenous meloxicam 5–15 mg, 30 mg and 60 mg and placebo groups, respectively. Investigators assessed hematoma events as mild (50%) or moderate (50%) in severity; each case resolved following intervention (heat therapy, phototherapy or incision and drainage). Four events (67%) were considered unrelated to study drug; two events (33%, wound hematomas in the intravenous meloxicam 30 mg group) were considered by the investigator to be possibly related to study medication.
Hemorrhagic events (defined as slight bleeding at incision site, postoperative incision bleeding, postprocedural hemorrhage, rectal bleeding due to constipation and vaginal spotting/bleeding after gynecological surgery) were reported for 3/517 (0.6%) subjects who received placebo, 3/910 (0.3%) subjects who received intravenous meloxicam 30 mg and 1/189 (0.5%) subjects who received intravenous meloxicam 60 mg. The four hemorrhagic events in the intravenous meloxicam groups were assessed as mild or moderate in severity; three resolved with no intervention; one resolved following ligation of an active vessel bleed and transfusion of 6 units of packed red cells. Two hemorrhagic events in the intravenous meloxicam group were considered by the investigator to be possibly related to study drug. One hemorrhagic event in the placebo group was severe and resolved after surgical evacuation of a hematoma and clipping of an active vessel bleed and infusion of 3 units of packed red cells. The investigator determined this event was possibly related to treatment. The other two hemorrhagic events in the placebo group were assessed by investigators as being mild in intensity and unrelated to treatment.
Cardiovascular events occurred in approximately 1% of subjects who received placebo and <1% of subjects who received intravenous meloxicam (table 4). Hypertension was the most commonly reported cardiovascular event (placebo, 4/517 [0.8%]; intravenous meloxicam 30 mg, 5/910 [0.5%]). All but one of these subjects had a prior medical history of hypertension. There were four reports of ECG abnormalities. Investigators assessed the severity of ECG abnormalities to be mild; none required treatment. Additional details describing ECG findings are provided in online supplemental digital appendix 1. One subject treated with placebo had myocardial ischemia secondary to coronary artery disease that led to prolonged hospitalization and was considered an SAE.
Injection site reactions were reported for approximately 1% of subjects (6/517, 1.2%) who received placebo and <1% of subjects (12/1426, 0.8%) who received intravenous meloxicam (table 4). Injection site reactions were assessed by the investigator to be mild (89%) or moderate (11%) in intensity; the reactions resolved without additional treatment. The most commonly reported injection site event was infusion/injection site pain, which occurred in 3/517 (0.6%) subjects who received placebo and 5/910 (0.5%) subjects who received intravenous meloxicam 30 mg. Phlebitis (including injection site phlebitis, vessel puncture site phlebitis and infusion site phlebitis) was reported for 1/517 (0.2%) subjects in the placebo group and 3/910 (0.3%) subjects in the intravenous meloxicam 30 mg group. Other events associated with the injection site (infusion site thrombosis, injection site erythema, injection site induration, infusion site pruritus and injection site infection) were reported for 4/517 (0.8%) subjects in the placebo group and 4/910 (0.4%) subjects in the intravenous meloxicam 30 mg group.
Hepatic events were reported for approximately 5% of subjects in the placebo group and 2%–5% of subjects in the intravenous meloxicam groups. Three subjects (placebo, n=1; intravenous meloxicam 30 mg, n=2) had severe events (‘liver function test abnormal’). Increased transaminases were reported at a higher rate in the intravenous meloxicam 30 mg group compared with the placebo group (placebo, 0/517 [0%]; intravenous meloxicam 30 mg, 2/910 [0.2%]).
Renal events were reported for <1% of subjects in the placebo and intravenous meloxicam groups (table 4). Investigators assessed two renal events in the intravenous meloxicam 30 mg group to be of moderate intensity; all other events were assessed as mild in intensity. Two subjects experienced an SAE of acute renal failure of moderate intensity; for the first subject, the renal event occurred on day 18 (the final meloxicam dose was given on day 4) and for the other subject, the event occurred on day 21 (the final meloxicam dose was given on day 3). In both cases, investigators considered the events to be unrelated to study medication.
Ketonuria was reported by one site in a single study (NCT01084161).4 Ketonuria occurred during the double-blind and open-label phases of the study and was reported for 5/64 (7.8%) subjects in the placebo group and for 23/211 (10.9%), 6/60 (10.0%), and 10/89 (11.2%) subjects in the intravenous meloxicam 5–15 mg, 30 mg and 60 mg groups, respectively.
Thrombotic events, including postprocedural pulmonary embolism and deep vein thrombosis, were reported for <1% of subjects in the placebo and intravenous meloxicam groups (table 4). A single subject (0.2%) in the placebo group experienced a thrombotic event (following abdominoplasty) as did four subjects (0.4%) in the intravenous meloxicam 30 mg group (following total knee replacement [n=3] and hernia repair [n=1]). One subject who received 2 doses of intravenous meloxicam 30 mg following total knee replacement surgery had a severe postprocedural pulmonary embolism/postoperative pulmonary embolus. The investigator assessed the event as unrelated to study drug. Of the remaining three events reported by subjects in the meloxicam group, none was considered by investigators to be related to study drug.
Wound healing events across the seven pooled studies were reported in <5% of subjects who received placebo or intravenous meloxicam (table 4). The rate of wound infection was ~1% and consistent across treatment groups (4/517 [0.8%], 3/327 [0.9%], 7/910 [0.8%] and 1/189 [0.5%] in the placebo, intravenous meloxicam 5–15 mg, intravenous meloxicam 30 mg and intravenous meloxicam 60 mg treatment groups, respectively). When ‘cellulitis’ and ‘incision site cellulitis’ were analyzed together, the rate of cellulitis associated with the wound was <1% in the placebo, intravenous meloxicam 30 mg and 60 mg groups (3/517 [0.6%], 8/910 [0.9%] and 1/189 [0.5%], respectively). When abscesses associated with wound healing (mesenteric abscess, periumbilical abscess, postoperative abscess and abdominal abscess) were analyzed collectively, the rate of wound healing abscesses was 2/517 (0.4%) in the placebo group and 2/910 (0.2%) in the intravenous meloxicam 30 mg group. Wound dehiscence was reported for 2/517 (0.4%), 1/327 (0.3%), 4/910 (0.4%) and 0/189 (0%) of the placebo, intravenous meloxicam 5–15 mg, intravenous meloxicam 30 mg and intravenous meloxicam 60 mg groups, respectively. In the pooled phase III studies, investigator satisfaction with wound healing was similar between intravenous meloxicam 30 mg and placebo at each postoperative evaluation. Mean assessment scores of 9.4/10 suggest that investigators were consistently satisfied with overall wound healing across groups. The incidence of clinically significant abnormal wound healing as determined by the investigator performing these evaluations was 9/393 (2.3%) for placebo and 16/748 (2.1%) for intravenous meloxicam 30 mg.
Other information relating to TEAEs
Within any of the treatment groups (placebo, intravenous meloxicam 5–15 mg, 30 mg and 60 mg), data did not indicate a greater incidence of TEAEs overall or in AEOSI, or acute reactions by total exposure, risk status, age, sex, race, history of select medical conditions (hypertension or diabetes) or the use of concomitant medications (antihypertensive or antithrombotic). Among subjects with a history of GI conditions, no data suggested a greater incidence of TEAEs overall, AEOSI or acute reactions apart from anemia discussed above.
Clinically meaningful abnormal laboratory results
Site investigators assessed results from laboratory tests, and any measures that were out of the range of normal and evaluated as clinically relevant were subsequently recorded as TEAEs. First, a higher percentage of subjects in the intravenous meloxicam 30 mg group had hemoglobin and hematocrit values that shifted from normal at baseline to low during the study compared with subjects in the placebo group (hemoglobin: 244/675 [36.1%] vs 98/346 [28.3%]; hematocrit: 259/663 [39.1%] vs 112/346 [32.4%]). Second, the decreases from baseline to the end of therapy in hematocrit and hemoglobin were greater in the intravenous meloxicam 30 mg group compared with the placebo group. At the end of study visit, hematocrit and hemoglobin trended toward, but did not reach, baseline in the intravenous meloxicam 30 mg group. In the placebo group, hematocrit and hemoglobin trends at the end of study were similar to the end of therapy. One subject randomized to placebo in study NCT02678286 and one subject randomized to intravenous meloxicam 30 mg in study NCT02720692 had a report of both ‘hematocrit decreased’ and ‘hemoglobin decreased’ that were deemed clinically significant and reported as an AE.
The only notable differences between the treatment groups in the percentage of subjects with potentially clinically significant chemistry laboratory parameters of special interest was a higher percentage of subjects in the intravenous meloxicam 30 mg group had albumin values that shifted from normal at baseline to low during the study compared with subjects in the placebo group (96/743 [12.9%] vs 17/390 [4.4%]). Although both intravenous meloxicam 30 mg and placebo groups had decreases from baseline to the end of treatment in albumin, the intravenous meloxicam 30 mg group had a greater shift away from baseline. By the end of study visit (approximately 7 days after the last dose), albumin approximated baseline in both groups. One report of ‘blood albumin decreased’ (mild intensity) in the intravenous meloxicam 30 mg group was deemed clinically significant and reported as a TEAE.
There were no notable differences between the treatment groups in the percentage of subjects with potentially clinically significant coagulation laboratory parameters of special interest (activated partial thromboplastin time, prothrombin time and prothrombin international normalized ratio). The incidence of clinically significant coagulation parameters reported as TEAEs was 4/748 (0.5%) and all occurred in the intravenous meloxicam 30 mg group; investigators assessed the events to be mild or moderate in intensity.
Changes in individual laboratory parameters over time were evaluated using shift plots. Among the subjects treated with intravenous meloxicam 30 mg, no individual laboratory parameter was identified as having a clinically meaningful shift.
Changes in vital signs
The incidence of clinically significant vital sign parameters reported as TEAEs was low and was generally similar between treatment groups in the pooled phase III studies. All events occurred only once. The most commonly reported vital sign TEAEs in the intravenous meloxicam 30 mg group were hypotension (11/748 [1.5%]) and fever (11/748 [1.5%]). Corresponding rates in the placebo group were 3/527 (2.0%) and 7/527 (1.8%), respectively.
Opioid consumption
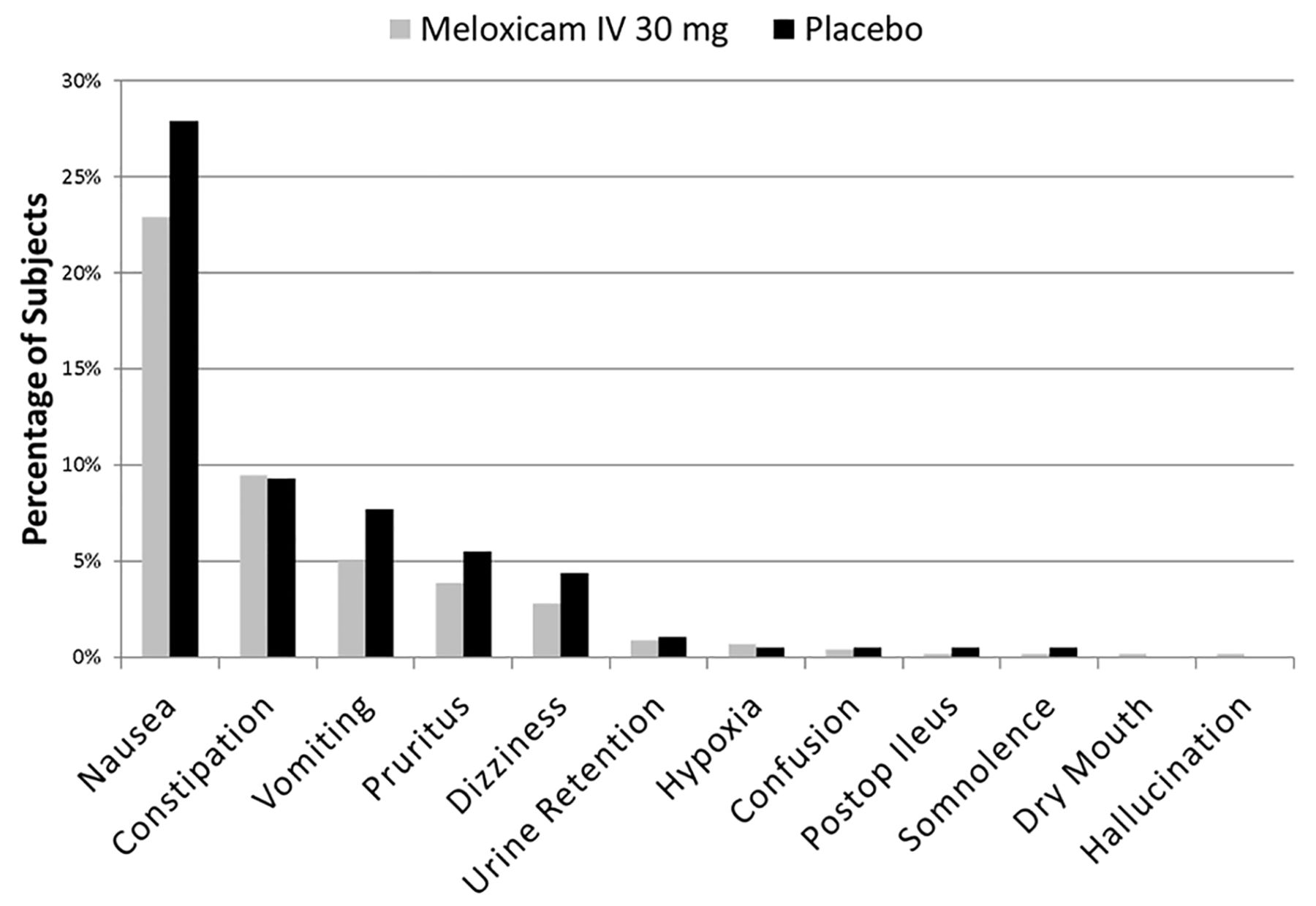
Mean opioid consumption was assessed in the phase III safety and tolerability study in subjects undergoing major surgery.9 Mean opioid consumption in the overall population was significantly less in the intravenous meloxicam 30 mg group compared with the placebo group in the hour 0‒24, hour 0‒48 and hour 0‒72 intervals (p<0.05) (table 7). Decreased opioid use among subjects treated with intravenous meloxicam 30 mg compared with placebo-treated subjects was observed across all subgroups (ie, surgery type, risk group and demographic characteristics). In the intravenous meloxicam 30 mg treatment group, decreased opioid use corresponded to fewer TEAEs commonly associated with opioid administration during the initial postoperative period (figure 1). Although a statistical evaluation was not conducted across the pooled studies, a trend indicating a decrease in adverse events commonly associated with opioid administration such as nausea, vomiting, constipation and pruritus was observed (tables 2 and 3).
Summary of total opioid consumption (mean±SD*) by time interval in subjects undergoing major surgery (NCT02720692)
{kind=link}
Treatment-emergent adverse events commonly associated with opioid use (NCT02720692).
Discussion
Intravenous meloxicam was generally well tolerated postoperatively in this pooled safety analysis. The most commonly reported treatment-related TEAEs across groups were nausea, vomiting and headache, all of which occurred with numerically lower frequency with intravenous meloxicam versus placebo. These findings are consistent with evidence from studies conducted with orally and rectally administered meloxicam in similar populations,12–15 with no new safety issues identified for the intravenous formulation.
In this analysis, TEAE rates were similar across populations, including older subjects (>65 years) with impaired renal function. Among subjects who received intravenous meloxicam 30 mg, constipation occurred at a higher rate in this subgroup compared with younger subjects with normal renal function (16.8% vs 5.5%, respectively). No other notable differences in TEAEs between subjects in these subgroups who received intravenous meloxicam 30 mg were reported. This finding is consistent with data from studies conducted with oral meloxicam, which concluded that there was no increased risk of toxicity among subjects with mild or moderate renal impairment relative to those having normal renal function.16 17
In this pooled analysis, bleeding events occurred in 2.1% of subjects in the placebo group and 3.6% of subjects in meloxicam treatment groups. The relatively high rates of bleeding events in the intravenous meloxicam 5–15 mg and 60 mg groups were largely driven by NCT01084161,4 where investigators were required to report any hemoglobin value ≤80.0 g/L after surgery as a TEAE (irrespective of clinical significance) as well as any hemoglobin value after surgery that was between 80.0 g/L and 90.9 g/L and had decreased by ≥5.0 g/L from screening. A post hoc analysis was performed to further examine the effect of treatment on hemoglobin; this analysis revealed no dose-related trends in the incidence of hemoglobin shifts from normal to low, and the difference between the shifts for active treatment and placebo was small enough that the effect of treatment on hemoglobin was considered similar to that of placebo. Pooled data from the phase III studies showed no notable differences between the treatment groups in the percentage of subjects with potentially clinically significant changes in coagulation parameters of special interest (activated partial thromboplastin time, prothrombin time and prothrombin international normalized ratio). Concomitant antithrombotic use was associated with proportional increases in rates of anemia (vs no antithrombotic use) in both the placebo and intravenous meloxicam 30 mg treatment groups. The incidence of bleeding events in this analysis is consistent with the published literature, including a meta-analysis of data from 16 postoperative studies that concluded that single doses or short courses of highly selective COX-2 inhibitors in the perioperative setting do not increase perioperative bleeding risk18 as well as data from meloxicam studies demonstrative of acceptable GI tolerability (low incidence of GI bleeding).19–21
Shift plots provided information regarding individual laboratory parameter changes over time. Although shifts in individual parameters were observed, the shifts were generally similar between subjects in the intravenous meloxicam 30 mg and placebo groups and were potentially attributable to the surgical procedures they had undergone.
In the pooled analysis, cardiovascular event rates were similar across treatment groups. The studies pooled in this report were of limited duration, which may have resulted in few observed significant differences in rates of cardiovascular events. Pooled data from the phase III studies showed that the incidence of clinically significant ECG parameters reported as TEAEs was low in both treatment groups. Data from phase II studies support these findings, as do published reports of the cardiovascular safety of oral meloxicam.12 22 One systematic review of data from 19 meloxicam studies (observational and randomized, controlled trials) concluded that meloxicam increased composite cardiovascular/renal risk slightly but did not elevate myocardial risk.22 A 2016 review of 40 published studies concluded that short-term NSAID use does not increase the risk for adverse cardiovascular, GI, renal or respiratory events in most subjects.23
Unexpected ketonuria events were reported across all treatment groups at a single site in NCT01084161,4 where parenteral acetaminophen was typically the first line of treatment for postoperative pain management rather than NSAIDs. This outcome may reflect local practices in the timing of resumption of adequate oral caloric intake and/or the use of glucose-containing electrolytes.4
Across the phase II/III clinical trial program, investigators followed good clinical practice with regard to reporting AEs. There were no uniform consensus definitions for reporting AEs across all studies. Difference in surveillance of AEs between studies is a limitation of this pooled analysis of safety findings.
In the phase II/III postsurgical study program where opioid rescue medication consumption was monitored, intravenous meloxicam was often associated with prolonged time to first rescue medication use3 4 6 7 and reduced rescue medication requirements.3 4 6 8 In both the phase II dental impaction and hysterectomy studies, the percentage of subjects using opioid medication after surgery was lower in the intravenous meloxicam 30 mg group compared with the placebo group (37% vs 95% and 58% vs 93%, respectively).3 4 The administration of intravenous meloxicam in the safety and tolerability study (NCT02720692)9 was associated with reduced opioid consumption (table 7). Many expert organizations now recommend strategies such as multimodal analgesia, which can include NSAIDs, to reduce excessive opioid use.11 24 25
Conclusions
The pooled safety data from the phase II/III clinical program demonstrate thatintravenous meloxicam was generally well tolerated in subjects with moderate to severe postoperative pain as indicated by a low incidence of TEAEs that was comparable with placebo. In trials where opioid use was monitored, meloxicam reduced postoperative rescue opioid use. These findings suggest that intravenous meloxicam may represent a useful alternative to current postoperative pain management options (non-selective NSAIDS and opioid analgesics). Additional studies are needed to define the optimal role for intravenous meloxicam in this setting.
Acknowledgments
Assistance with manuscript preparation was provided by Mary Tom, PharmD, and Susan Martin, PhD, of The Medicine Group (New Hope, PA), and was funded by Recro Pharma, Inc. (Malvern, PA).
References
Footnotes
Funding Funding for this research was provided by Recro Pharma, Inc., Malvern, PA.
Competing interests ERV has received consulting fees from Cara, Mallinckrodt Pharmaceuticals, Merck, Recro, Salix Pharmaceuticals, Inc and Trevena, Inc and has received grants and consulting fees from AcelRx Pharmaceuticals, Inc, Durect and Pacira Pharmaceuticals, Inc, outside this submitted work. TJG has received consulting fees from Edwards, Mallinckrodt, Merck, Recro and Medtronic, Inc. RJM, SWM and SH are employees of Recro Pharma, Inc, Malvern, Pennsylvania. WD receives consultancy fees from Recro Pharma, Inc, Malvern, Pennsylvania. SB is an employee of Ohio State University, which participated in trial NCT02720692. NS is an employee of Lotus Clinical Research, which participated in trials NCT02678286, NCT02675907 and NCT02720692 as a CRO and/or research site; NS and Lotus have also received study grants and other compensation for clinical trial services from multiple pharmaceutical companies.
Patient consent for publication Not required.
Ethics approval Institutional Review Board.
Provenance and peer review Not commissioned; externally peer reviewed.