Article Text

Abstract
The blood–brain barrier (BBB) describes the unique properties of endothelial cells (ECs) that line the central nervous system (CNS) microvasculature. The BBB supports CNS homeostasis via EC-associated transport of ions, nutrients, proteins and waste products between the brain and blood. These transport mechanisms also serve as physiological barriers to pathogens, toxins and xenobiotics to prevent them from contacting neural tissue. The mechanisms that govern BBB permeability pose a challenge to drug design for CNS disorders, including pain, but can be exploited to limit the effects of a drug to the periphery, as in the design of the peripherally acting μ-opioid receptor antagonists (PAMORAs) used to treat opioid-induced constipation. Here, we describe BBB physiology, drug properties that affect BBB penetrance and how data from randomized clinical trials of PAMORAs improve our understanding of BBB permeability.
- pharmacology: additive agents
- pharmacology: opioids
- opioids, adverse effects
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, an indication of whether changes were made, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Maintenance of the central nervous system (CNS) environment is important for its function and is accomplished via three protective layers that partition the blood from neural tissue. The choroid plexus epithelium forms the blood–cerebral spinal fluid (CSF) barrier by secreting CSF into the cerebral ventricles, while the arachnoid epithelium separates the blood from the subarachnoid CSF.1–3 The blood–brain barrier (BBB) is the third structure that separates the blood and neural tissue, and refers to the unique properties of the endothelial cells (ECs) that line the microvasculature of the CNS.4 Here, we describe contributions of the BBB to CNS homeostasis, particularly its differential permeability to drugs and other substances. We discuss the pharmacology of peripherally acting μ-opioid receptor antagonists (PAMORAs), whose design capitalizes on this differential permeability.
Physiology of the BBB
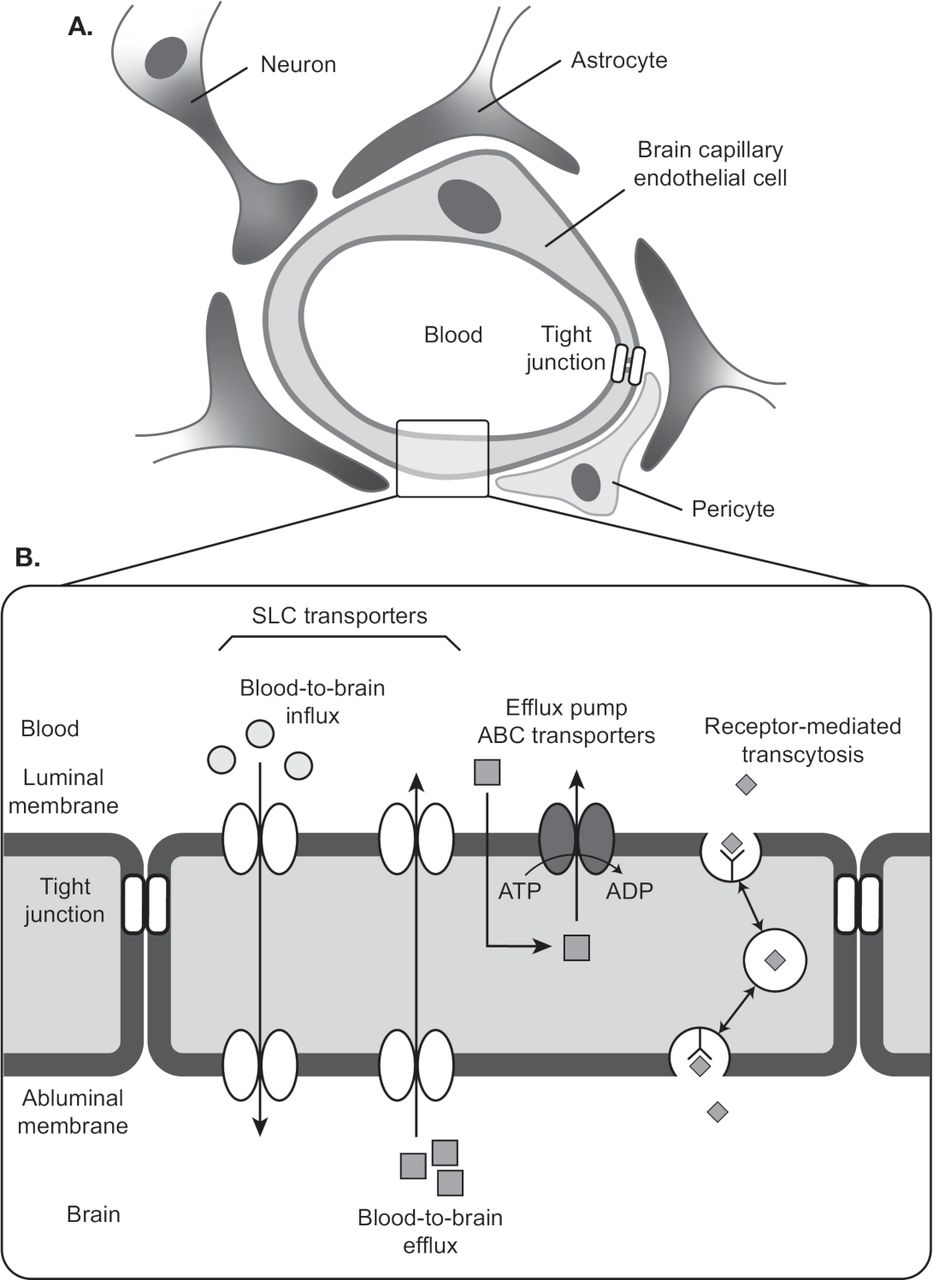
Blood vessels are composed of ECs and mural cells (ie, vascular smooth muscle cells and pericytes; figure 1A). The diverse functions of the BBB are mediated primarily by ECs, which form the walls of capillaries and have unique properties compared with ECs in other tissues.4–11 ECs of the BBB have a greater concentration of mitochondria, which may be related to the energy required to maintain ion gradients necessary for transport functions.12 ECs of the BBB also have fewer pinocytotic vessels than ECs found elsewhere, which results in a low level of passive solute movement out of the bloodstream and into the interstitial fluid.9 Finally, very low levels of leukocyte adhesion molecules in ECs of the BBB restrict inflammatory immune cells from entering the CNS.4
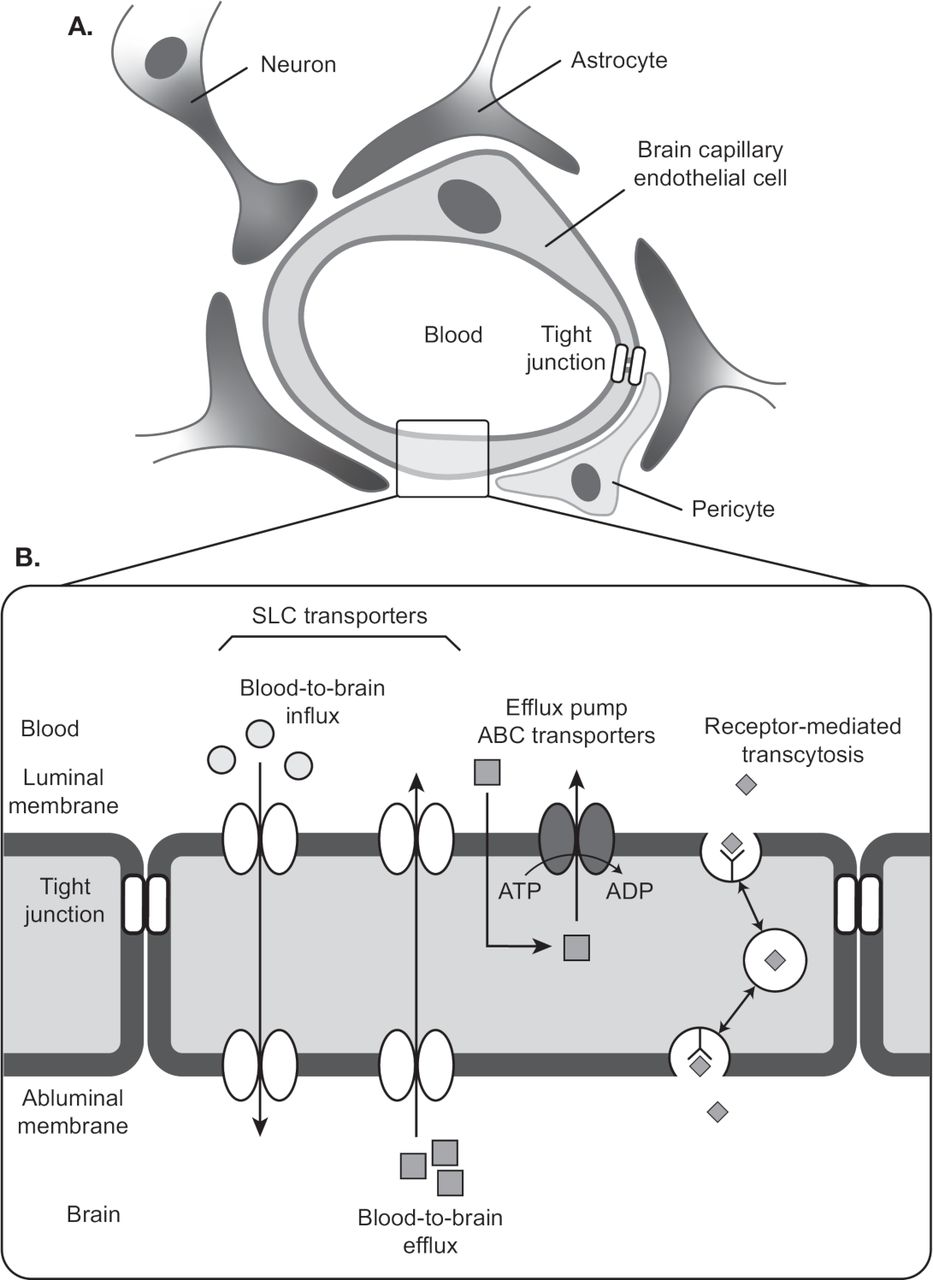
Cellular and structural components of the blood–brain barrier. (A) A single layer of ECs line the capillaries of the brain’s vasculature. ECs are connected by a network of tight junctions that inhibit paracellular transport. Pericytes and astrocytes provide mechanical and functional support to the BBB by ensheathing ECs at the abluminal walls. (B) Transcellular transport of nutrients and waste is mediated by solute carrier transporters. ABC transporter is an efflux pump that pumps drugs from ECs to blood. Receptor-mediated transcytosis mediates transport of macromolecules such as insulin across the BBB. Adapted from Ohtsuki et al.19 Quantitative targeted proteomics for understanding the blood-brain barrier: towards pharmacoproteomics. Expert Rev Proteomics. 2014;11:303–313. Reprinted by permssion of the publisher (Taylor & Francis, Ltd, http://www.tandfonline.com). ABC, ATP-binding cassette; BBB, blood–brain barrier; EC, endothelial cell; SLC, solute carrier.
ECs of the BBB rely on tight junctions (TJs) and an assortment of transport proteins to control movement of substrates into and out of the capillary lumen.4 6–8 10 11 TJs are multiprotein complexes that connect neighboring ECs via their lateral membranes. They consist of transmembrane proteins of the claudin and occludin families. Protein–protein interactions within and between cells create the functional core of the TJ, which has a central pore of a few nanometers. The TJ pore will allow transport of some small molecules, but paracellular transport between ECs is generally constrained. The core proteins of the TJ associate with membrane-associated cytoskeletal proteins and adaptor proteins that may regulate TJ stability and permeability.4 TJs are stabilized by the glia limitans, a basement membrane formed by astrocytic end-feet, and by astrocyte-mediated signaling to ECs.2 4 Astrocytes also contribute to BBB health by metabolizing drugs or other toxic compounds that leave the ECs.13
While TJs regulate paracellular movement between blood and brain, ECs are equipped with efflux pumps and solute carrier transporters that mediate transcellular passage (figure 1B). Because TJs create well-separated luminal and abluminal domains, the directionality of transcellular movement for many substances is determined by the domain in which the transporter is located.14 Efflux pumps in the luminal membrane of the ECs move many small lipophilic molecules out of the EC cytoplasm into the blood, thus limiting their passive diffusion into the CNS. Among the best-characterized efflux pumps are members of the ATP-binding cassette (ABC) transporter family: the P-glycoprotein transporter (P-gp),15 multidrug resistance-associated proteins (MRP),16 and breast cancer resistance protein.17 18 Solute carrier transporters deliver glucose, large amino acids, and other nutrients across the EC from blood to brain, and eliminate metabolic waste products, glutamate, and toxins via transport from brain to blood.19 20 Receptor-mediated transcytosis is another means by which proteins such as insulin are moved across ECs of the BBB.19
Similar to astrocytes, pericytes provide mechanical and functional support to the BBB by ensheathing ECs at the abluminal wall within the basement membrane.4 21 Pericytes form peg–socket contacts with ECs that allow for the exchange of ions, metabolites, second messengers, and ribonucleic acids between the 2 cell types.21 Evidence suggests that pericytes aid BBB stability via their roles in angiogenesis, deposition of extracellular matrix, and blood flow response to neural activity.4 21 Coordinated paracrine interactions between ECs and the pericytes, astrocytes, microglia and neurons that surround them form the ‘neurovascular unit’, whose disruption accompanies cerebrovascular disease.22
Determinants of BBB permeability
To ensure proper function and maintain the homeostatic environment of neural tissue, the BBB must closely regulate entry to and exit from the CNS. This includes not only endogenous molecules such as peptides, glucose and other nutrients, and waste products, but also therapeutic medications. Studies of drug databases indicate that only ~10% of drugs are CNS active.23 The BBB maintains its highly restrictive nature via passive and active mechanisms. Drugs that successfully use passive diffusion to traverse the lipid bilayer of the BBB fulfill several criteria related to their intrinsic and biochemical properties.23–28 One of the intrinsic properties governing permeability is molecular weight. Molecules that weigh more than 400 Da are generally not able to access the brain by crossing the BBB,23 29 although some larger molecules in the range of 500–600 Da are not excluded.25 30 Another determinant of permeability is molecular volume, which affects permeability in a non-linear manner. Fisher et al found a 100-fold decrease in penetration between molecules with volumes of 100 Å vs 50 Å.23 31 A compound’s hydrophilicity is the third major determinant of permeability. Water-insolvent substances that form seven or fewer bonds with water are thought to be able to diffuse through the lipid bilayers of ECs to reach the brain.23 32–34
While lipid insolubility prevents many drugs from reaching the brain via passive diffusion, active transport by the BBB’s efflux pumps is equally important to maintain the brain’s integrity. The P-gp transporter is characterized by a broad substrate specificity that is made possible by aromatic, polar, and non-polar residues within the substrate binding domains.35 The presence of multiple substrate binding sites that are both overlapping and nonoverlapping ensures that P-gp transporters at the BBB function efficiently without reaching saturation.28 36 37 Most substrates that interact with P-gp are weakly amphipathic and relatively hydrophobic, and are able to enter cells through passive diffusion across the cellular membrane.38 P-gp intercepts substrates within the membrane before they enter the cytosol and pumps them into the extracellular space via an energy-dependent transport cycle driven by ATP hydrolysis.38 39 P-gp can thus prevent diffusion of drugs to the brain and, more globally, can alter the clinical efficacy of the drugs it interacts with by altering their absorption and tissue distribution. Additionally, for drugs that are substrates of P-gp and rely on the interaction with P-gp for exclusion from the brain, exposure to P-gp inhibitors or conditions that cause P-gp deficiency can allow increased entry into the brain.
Unlike drug–receptor interactions, interactions between a drug and the BBB involve multiple components that contribute positively or negatively to its permeability. As discussed previously, the impact of the EC membrane varies with the degree of lipophilicity of the drug, being negative or positive, while the ABC efflux transporters have a negative effect. Besides these, the many solute carrier proteins present at the BBB will increase the permeability of any drug that can bind to them.14 Because a drug’s permeability profile is influenced by multiple components, subtle changes in structure can yield substantial changes in permeability, and drugs with similar functions do not penetrate the BBB equally. The comparison of morphine and codeine is an example of how the BBB shows differential permeability to structurally similar drugs. Although codeine might be expected to display a poorer permeability profile than morphine because of the methyl side chain, it penetrates the brain much more quickly (single-pass clearance of drug as a percentage of a simultaneously injected tritiated water internal standard: morphine, 2.6%±0.2%; codeine, 26.0%±2.0%).40–42 BBB penetrance of morphine and its active metabolites is governed by all three factors discussed here.43 Morphine is able to diffuse through the EC membrane, and it also reaches the brain via active transport; however, it is subject to efflux via MRP and P-gp.44–49 In contrast, codeine is not a substrate for MRP or P-gp and easily diffuses across the BBB.48–50 Table 1 presents a summary of the permeability profiles for morphine, codeine and other currently marketed opioid receptor agonists and antagonists.
Summary of permeability profiles for currently marketed opioid receptor agonists and antagonists
Causes of BBB dysfunction
When the BBB is compromised by stroke, total brain injury (TBI), or normal aging, patterns of BBB permeability are altered. Cerebral damage from ischemic stroke is generated immediately after the event and further develops over the next few days. This biphasic nature of injury, which can depend on the degree of hypoxia, occurs not only in the grey and white matter but also at the BBB. Studies in animal models suggest that as early as a few hours after a stroke, the permeability of the BBB increases. Further changes in permeability are detected days later.4 51 52
The response of the BBB to TBI has several similarities to the response to stroke. First, the timeline of response is biphasic, with the earliest changes observed in hours, and delayed-onset disruptions in permeability over days.53–58 Second, the abnormalities that are observed are similar, with upregulation of transcytosis.59 60 However, TBI results in extensive physical disruption of the vasculature, as some studies have reported localized swelling or constriction, ectasia, and membrane thickening of both capillaries and larger vessels.54 55
Aging, unlike stroke and TBI, results in uniform changes in the BBB across brain regions. Age-related BBB dysfunction is compounded by diminished function of the choroid plexus, which produces less CSF and transports less materials out of the ventricles.61–63 In the cerebrovasculature, blood flow and blood volume, and the corresponding level of oxygenation, are diminished in brains of older versus younger persons,64 and some studies have reported changes in capillary structure and density.65 The ECs of older individuals show alterations in their most highly specialized adaptations. These changes include reduced EC mitochondrial density, reduced capacity to transport waste from brain to blood, accumulation of extracellular matrix components and stiffening of the vessel wall, and loosening of TJs. Furthermore, a loss of pericytes compromises BBB integrity and causes hypoperfusion and secondary neurodegeneration.66 As a result, the aged have a generally leaky BBB, with increased permeability to many substances. Recent studies in humans and in animal models have reported decreased expression of P-gp as a result of aging.67 68 Therefore, a heightened response to many opioid medications would most likely be observed in both healthy and infirm older people. These results could potentially affect the tissue distribution of drugs such as PAMORAs that rely, at least partially, on P-gp activity to restrict its diffusion to the brain. Potential effects of aging and other causes of BBB compromise on CNS exposure to PAMORAs have not been extensively explored to date but are currently under investigation.
Peripheral opioid antagonism
One of the advantages of a highly impermeable BBB is that it can be exploited to limit the activity of a drug to the periphery. This strategy was used to develop PAMORAs,69 which are prescribed primarily for opioid-induced constipation (OIC). OIC frequently occurs with opioid use and is caused by activation of opioid receptors in the gastrointestinal tract. Signaling from μ-opioid receptors in the mucosal epithelium results in inhibition of neurons of the myenteric plexus, a key regulator of gastrointestinal motility (figure 2).69–75 Although many patients develop tolerance to opioid analgesia and require increasing doses for pain relief, little tolerance to OIC develops over time.76

Mechanism of peripherally acting µ-opioid receptor antagonists.69 Reprinted from The Lancet, 373, Becker G, Blum HE, Novel opioid antagonists for opioid-induced bowel dysfunction and postoperative ileus, 1198–1206, Copyright 2009, with permission from Elsevier. CNS, central nervous system.
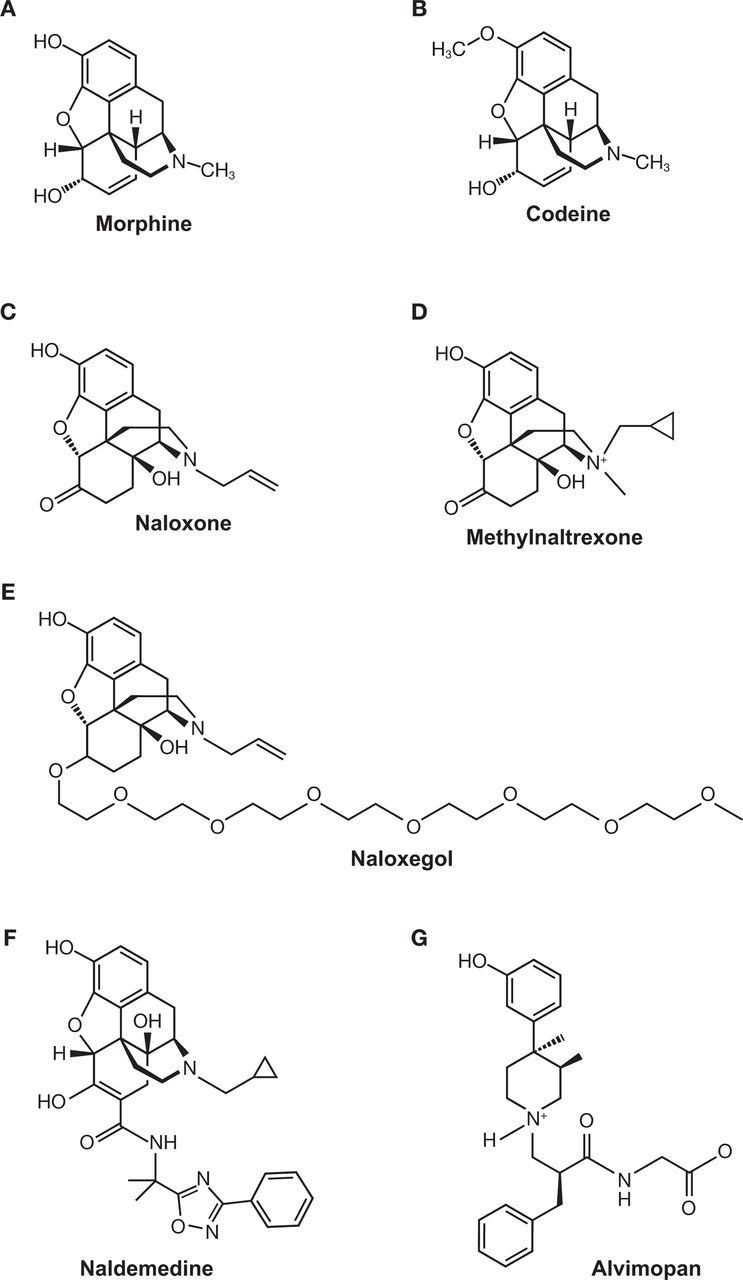
While laxatives are often the first treatment option for OIC, they frequently yield complications and poor outcomes.77–79 OIC can be also improved by use of opioid antagonists such as naloxone80 81; however, naloxone crosses the BBB and decreases opioid analgesia.72 82–84 PAMORAs are able to reduce the symptoms of OIC while maintaining the efficacy of opioid agonists in the CNS.73 76 85 86 Three PAMORAs have been developed to treat OIC: methylnaltrexone (Relistor),87 naloxegol (Movantik),88 and naldemedine (Symproic).89 A fourth PAMORA, alvimopan (Entereg), is indicated to accelerate the time to upper and lower gastrointestinal recovery following surgeries that include partial bowel resection with primary anastomosis.90 These PAMORAs exhibit distinct clinical profiles and employ different mechanisms that restrict their movement across the BBB. The chemical structures of each PAMORA, as well as the chemical structures for morphine, codeine and naloxone, for reference, are depicted in figure 3. Comparison of efficacy among the PAMORAs is difficult because clinical outcomes were not the same in all trials. However, there are several publications that have reviewed the efficacy and safety of this class.73 91–94
{kind=link}
{kind=link}
{kind=link}
Chemical structures of (A) morphine, (B) codeine, (C) naloxone, (D) methylnaltrexone, (E) naloxegol, (F) naldemedine, and (G) alvimopan.
One of the first PAMORAs to be developed was methylnaltrexone. As the name suggests, this drug is a modified form of naltrexone, with a methyl group added to the compound’s sole nitrogen. Because the resulting quaternary amine on methylnaltrexone is too polar to cross the BBB, exposure is predicted to be limited to peripheral receptors, including those in the gastrointestinal tract.87 95–97 Methylnaltrexone and its metabolites are not substrates for P-gp.
Methylnaltrexone was approved by the US Food and Drug Administration (FDA) in 2008 as an injection, and in 2016 as a tablet formulation. Both formulations are approved to treat OIC in adults with chronic non-cancer pain (CNCP). The injectable form is additionally approved for the treatment of OIC in adults with advanced illnesses or pain caused by active cancer who require opioid dosage escalation for palliative care.87 Multiple clinical trials conducted over the past 10 years have examined the maintenance of analgesia and the degree of withdrawal among patients taking methylnaltrexone for OIC,98–102 and these endpoints are key in vivo indicators of the ability of the BBB to restrict entry of methylnaltrexone. Two of these trials, reported by Webster et al in 2015 and Webster and Israel in 2018, examined analgesia and withdrawal by assessing the patients’ median morphine equivalent dose (MED) in addition to their pain intensity and symptoms of withdrawal as measured by the Subjective Opioid Withdrawal Scale and/or the Objective Opioid Withdrawal Scale. For patients receiving a 150, 300, or 400 mg oral dose of methylnaltrexone QD, the mean MED remained stable throughout 4 weeks of QD dosing and 8 weeks of as-needed (PRN) dosing.98 Among patients who received a 12 mg subcutaneous dose QD or QOD during 48 weeks of open-label treatment, the median daily MED also showed little variation (12 mg QD range: 150.0–180.0 mg/day; 12 mg QOD range: 144.0–162.6 mg/day).99 The authors also found no statistically significant difference in scores for pain intensity or withdrawal after 2 or 4 weeks of treatment with either the oral or injectable formulation.98 99 Two additional randomized methylnaltrexone trials examined analgesia and withdrawal by assessing the patients’ pain intensity and symptoms of withdrawal as measured by the modified Himmelsbach Scale.101 102 In both studies, patients with advanced illnesses and OIC received subcutaneous methylnaltrexone or placebo. During the double-blind phase of the study by Slatkin et al, patients received a single subcutaneous dose of 0.15 mg/kg methylnaltrexone, 0.30 mg/kg methylnaltrexone, or placebo.101 In the study by Thomas et al, patients received subcutaneous methylnaltrexone 0.15 mg/kg or placebo every other day for 2 weeks.102 No significant changes from baseline in pain scores or modified Himmelsbach scale scores were demonstrated at any time point in either study. Similar results have been observed in earlier trials of methylnaltrexone administered intravenously at doses of up to 24 mg every 6 hours103 and 0.3 mg/kg every 6 hours.104 Even with the high intravenous doses administered in these trials, signs of opioid withdrawal103 and pain intensity104 were similar to placebo.
The FDA approved another PAMORA derived from naltrexone. Naldemedine, approved in 2017, consists of a naltrexone backbone with a large steric side chain added at the ketone group. The side chain renders naldemedine highly polar, and with a molecular weight of 742.84 Da, it is unlikely to cross the BBB.89 105 Unlike methylnaltrexone, naldemedine is a substrate for P-gp.87 89 In pharmacokinetic studies, a single dose of the P-gp inhibitor cyclosporine resulted in mild increases in naldemedine Cmax and area under the plasma concentration time curve (AUC) (1.45-fold increase and 1.78-fold increase, respectively).89 A recent animal study suggested that while naldemedine is a substrate for P-gp, it is primarily excluded from the brain due to its limited ability to cross the BBB rather than efflux by P-gp.106
Oral naldemedine is indicated only for patients with OIC and CNCP.89 The evidence for naldemedine’s lack of central penetration was gathered from four clinical trials.107–110 The COMPOSE trials were phase III trials evaluating 0.2 mg daily naldemedine over 2 weeks of randomized treatment (COMPOSE-4), 12 weeks of randomized (COMPOSE-1 and COMPOSE-2) or open-label treatment (COMPOSE-5), or 52 weeks of randomized treatment (COMPOSE-3).108 109 111 All of the COMPOSE trials assessed withdrawal and the maintenance of analgesia. Over 12 weeks of treatment, scores on the Clinical Opioid Withdrawal Scale (possible scores, 0–48) showed little variation and were generally less than 1.0, with no statistically significant between-group differences in COMPOSE-1, COMPOSE-2, and COMPOSE-5. Scores on the pain numerical rating scale and the total weekly opioid dose (assessed in COMPOSE-1 and COMPOSE-2) were also similar.108 111 A 4-week randomized study by Webster et al confirmed that patients receiving naldemedine had levels of pain and withdrawal that were similar to those receiving placebo.110 The results of the COMPOSE-3 study also demonstrated that long-term use of naldemedine did not result in opioid withdrawal or decreased analgesia.109
Prior to approval of naldemedine in 2017, the FDA approved a PAMORA derived from naloxone. Naloxegol, approved in 2014, differs from naloxone by the addition of a PEG side chain.88 Like naldemedine, naloxegol is thought to be restricted from crossing the BBB because of its size (742 Da), its polar nature, and the activity of P-gp.88 Also like naldemedine, the drug’s pharmacokinetics can be influenced by treatment with P-gp inhibitors; when coadministered with the strong P-gp inhibitor ketoconazole, naloxegol Cmax and AUC increased 9.6-fold and 12.8-fold, respectively. Coadministration with the weak P-gp inhibitor quinidine increased naloxegol Cmax and AUC 2.5-fold and 1.4-fold, respectively.88 Naloxegol is manufactured as a tablet, with a dosage of 12.5 or 25 mg QD indicated for patients with OIC and chronic pain.88 Evidence of the lack of CNS penetration of naloxegol has been generated by clinical studies in patients with OIC and in healthy volunteers.112–116 A crossover, ascending dose study of naloxegol, conducted by Eldon et al, determined the pharmacokinetic and clinical properties of 8–1000 mg of daily naloxegol in healthy adult men. The authors reported that pupillary miosis, a readout of centrally acting morphine, was intact at doses up to 125 mg.112 Naloxegol’s performance in patients with OIC and chronic pain was evaluated in the phase III KODIAC trials (KODIAC-04, KODIAC-05, KODIAC-07, and KODIAC-08).113–116 Patients enrolled in two 12-week randomized studies (KODIAC-04 and KODIAC-05)115 and one 12-week randomized extension study (KODIAC-07)116 who received either daily placebo, 12.5 mg naloxegol, or 25 mg naloxegol experienced statistically similar levels of both worst and average pain and showed similar patterns of their daily doses of opioid medications.113 115 116 The percentages of patients who experienced no change in symptoms of withdrawal were similar among the treatment groups (72%–85%), and increased to >91% for all groups in the extension study.113 116 In the 52-week KODIAC-08 trial, the only one to compare naloxegol (25 mg QD) to an investigator-chosen laxative regimen, patients also reported similar levels of pain and daily opioid doses.114 However, it has been reported that a higher frequency of gastrointestinal adverse events potentially related to opioid withdrawal were observed in naloxegol-treated patients receiving methadone compared with other opioids for pain management.88 Additionally, possible opioid withdrawal, defined as at least three adverse reactions potentially related to opioid withdrawal that occurred on the same day and were not all related to the gastrointestinal system, was noted in 3% of patients who received naloxegol 25 mg vs 1% of patients who received naloxegol 12.5 mg and <1% of patients who received placebo in two studies regardless of maintenance opioid treatment.88 Collectively, the data indicate that the BBB functions as it should when naltrexone-based or naloxone-based PAMORAs are prescribed to patients with advanced illness or chronic pain. However, the package inserts for all of these opioid antagonists warn of complications of withdrawal among those with a leaky BBB.87–89
Alvimopan was approved in 2008 as a 12 mg capsule and is indicated for short-term, in-hospital use to accelerate gastrointestinal recovery after surgical bowel resection with primary anastomosis.90 Like other PAMORAs, it displays a highly preferential distribution to peripheral opioid receptors.117 The BBB restricts passage of alvimopan because an N-substituted side chain introduces a zwitterion, which renders the molecule large and highly lipophobic.90 117 In vitro studies suggest that alvimopan and its metabolite are substrates for P-gp; however, coadministration of mild-to-moderate P-gp inhibitors has not been demonstrated to influence alvimopan pharmacokinetic parameters, and clinical studies examining coadministration of a strong P-gp inhibitor have not been conducted.90Alvimopan is associated with a risk evaluation and mitigation strategy because patients in a 12-month trial of 0.5 mg two times per day alvimopan experienced a higher rate of myocardial infarction than did those in placebo groups.90 Although several phase III trials were undertaken to examine alvimopan’s effectiveness to treat OIC,118 development for OIC was discontinued due to a lack of consistent efficacy data among patients with chronic pain.76 119–121
Conclusion
The selective permeability of the BBB is integral to the maintenance of CNS homeostasis. Although the permeability of the BBB is resistant to manipulation, it can be used as a tool to restrict the activity of a drug to the periphery. The development of PAMORAs to treat OIC illustrates the success of this approach, which has tremendous implications for drug design. However, the potential of a permeability-driven approach can only be realized when we have a thorough understanding of how the BBB is disrupted in the most common neurological conditions.
References
Footnotes
Contributors Technical editorial and medical writing assistance was provided under the direction of the authors by Dana A. Franznick, PharmD, of Echelon Brand Communications, LLC, an OPEN Health company, Parsippany, New Jersey, USA.
Funding Funding for this assistance was provided by Bausch Health LLC, USA.
Competing interests ERV has received compensation for consulting from AcelRx, Avenue, Cara, Concentric, Heron, Innocoll, Mallinckrodt, Merck, Neumentum, Pacira, Pfizer, Recro, Salix, and Trevena, and has received grants from Pacira and Durect. ARV has nothing to disclose.
Patient consent for publication Not required.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement All data relevant to the study are included in the article or uploaded as supplementary information.