Article Text

Abstract
Introduction Peripheral nerve stimulation (PNS) has historically been used to treat chronic pain, but generally requires implantation of a permanent system for sustained relief. A recent study found that a 60-day PNS treatment decreases post-amputation pain, and the current work investigates longer-term outcomes out to 12 months in the same cohort.
Methods As previously reported, 28 traumatic lower extremity amputees with residual and/or phantom limb pain were randomized to receive 8 weeks of PNS (group 1) or 4 weeks of placebo followed by a crossover 4 weeks of PNS (group 2). Percutaneous leads were implanted under ultrasound guidance targeting the femoral and sciatic nerves. During follow-up, changes in average pain and pain interference were assessed using the Brief Pain Inventory–Short Form and comparing with baseline.
Results Significantly more participants in group 1 reported ≥50% reductions in average weekly pain at 12 months (67%, 6/9) compared with group 2 at the end of the placebo period (0%, 0/14, p=0.001). Similarly, 56% (5/9) of participants in group 1 reported ≥50% reductions in pain interference at 12 months, compared with 2/13 (15%, p=0.074) in group 2 at crossover. Reductions in depression were also statistically significantly greater at 12 months in group 1 compared with group 2 at crossover.
Conclusions This work suggests that percutaneous PNS delivered over a 60-day period may provide significant carry-over effects including pain relief, potentially avoiding the need for a permanently implanted system while enabling improved function in patients with chronic pain.
Trial registration number NCT01996254.
- peripheral nerve stimulation
- neuropathic pain
- phantom pain
- post-amputation pain
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, an indication of whether changes were made, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Chronic pain is a prevalent and costly condition estimated to occur in over 30% of the population.1 Chronic pain frequently causes disability and depression, and has a strong negative correlation with qualify of life.2 Peripheral nerve stimulation (PNS) has been successful in treating many chronic pain conditions,3 including nerve injury, complex regional pain syndrome, occipital neuralgia, and post-surgical pain, but conventional PNS has historically been limited by invasiveness and complications.4
Conventional neurostimulation systems, including PNS and spinal cord stimulation (SCS), have been found to produce sustained pain relief when implanted and used long term. However, opportunities have been rare to study the sustained effects of limited durations of stimulation on chronic, intractable pain. Patients with favorable responses during short-term stimulation trials typically progress to system implantation, and patients who have implanted systems deactivated or explanted often do so because of loss of therapeutic effect or complications like hardware failures.4 In studies evaluating the carry-over effects of PNS or SCS, only short stimulation periods (minutes to hours) were tested, and Wolter et al noted that there are no data on the duration of post‐SCS pain relief in humans.5 6
A novel PNS system that is now Food and Drug Administration (FDA)-cleared delivers stimulation percutaneously for up to 60 days before the removal of the stimulating leads.7 8 The system was designed to overcome many of the limitations of conventional PNS systems, aiming to reduce invasiveness by placing leads percutaneously (typically with ultrasound guidance) and reduce infection rates and the loss of therapeutic effect due to lead migration by employing a flexible, open-coil lead placed remote from the target nerve.7–10 The development of such a system has enabled the evaluation of long-term relief of pain following a PNS treatment that significantly exceeds the average length of a percutaneous stimulation trial (4–7 days).
A multicenter, randomized, double-blind, placebo-controlled trial was conducted to evaluate changes in chronic pain and functional outcomes following amputation up to 12 months after a 60-day PNS treatment. Post-amputation pain, including residual limb pain (RLP) and phantom limb pain (PLP), is commonly neuropathic in origin, though RLP may also have non-neuropathic etiologies.11 The primary endpoint of the study was previously reported to be positive, finding that a greater proportion of participants receiving 4 weeks of PNS reported substantial (≥50%) reductions in pain compared with those receiving 4 weeks of placebo to a statistically significant degree.12 The current work presents the long-term follow-up of the study cohort through 12 months, prospectively evaluating secondary outcomes including sustained reductions in pain, pain interference, and depression, and patient-reported global improvement, and exploring the unique mechanisms that may underlie the maintenance of pain relief following the 60-day treatment.
Materials and Methods
Study design and population
This multicenter, randomized, double-blind, placebo-controlled, partial-crossover study was designed to collect data regarding the safety and effectiveness of percutaneous PNS for chronic pain in amputees. The study was approved by the FDA under an Investigational Device Exemption, and the PNS system is now FDA-cleared.
Prospective participants at six enrolling centers in the USA provided written consent and were assessed for eligibility. Participants were traumatic lower extremity amputees ≥18 years of age with moderate-to-severe (≥4 on a 0–10 pain rating scale13) RLP and/or PLP. Other than requiring a healed residual limb without comorbidities, there were no constraints on time since amputation. Key exclusion criteria included changes in pain medications in the previous 4 weeks, Beck Depression Inventory II (BDI-II) score >20, compromised immune system (eg, HIV, chemotherapy, immunosuppressive medications), diabetes mellitus type I or II, implanted electrical stimulation devices, anticoagulation therapy (aside from aspirin or warfarin with an international normalized ratio (INR) ≤1.5), history of a bleeding disorder, history of valvular heart disease, pregnancy, confounding central nervous system (CNS) disorders, allergy to local anesthetic agents or skin-contact materials, history of recurrent skin infection, botulinum toxin injection in the previous 3 months in the affected limb, and steroid injection in the previous 6 weeks in the affected limb. Continued use of all baseline pain medications was permitted at or below the documented baseline dosages.
A 7-day baseline pain diary was completed to determine average daily RLP and PLP scores and document baseline dosage of pain medications. Qualifying participants were randomized 1:1 in blocks of two to one of two groups, stratified by enrolling institution, using a masked allocation sequence generated by the study’s data capture system. Treating physicians were unmasked, while participants and outcomes assessors were masked to group assignment. Group 1 (PNS) received active stimulation for 4 weeks, while group 2 (placebo) received sham stimulation. After the first 4 weeks, group 1 received four additional weeks of stimulation (up to 60 days total) and group 2 crossed over to receive active stimulation for 4 weeks. After the 8-week treatment period, all leads were removed and both groups were followed monthly for up to 12 months from the initial implantation.
Interventions
Percutaneous PNS leads were implanted under ultrasound guidance targeting the femoral and sciatic nerves.10 Each introducer entry site was prepped using aseptic technique, and cutaneous local anesthesia was administered, taking care to not deliver anesthetic to the deeper target nerve where it may affect the stimulation response. The femoral nerve was targeted with the participant supine using a lateral approach approximately 1–2 cm distal to the inguinal crease. The lead was implanted remote (0.5–3 cm) from the nerve to enable selective activation of large-diameter sensory fibers.7 8 10 The sciatic nerve was targeted with the participant prone or in the lateral decubitus position. Ultrasonic landmarks included the greater trochanter, ischial tuberosity, femur, and/or popliteal artery to guide location of the lead remote from the nerve proximal to the level of amputation. Modifications to these approaches were made at the discretion of the investigator based on patient-specific anatomy.
In the PNS group, a monopolar needle electrode was typically inserted to within 0.5–3 cm of the targeted nerve. Test stimulation (asymmetric charge-balanced biphasic pulse train, 100 Hz, 1–30 mA, 10–200 µs) was delivered to confirm that comfortable stimulation-evoked sensations could be induced in the regions of RLP and/or PLP. The test needle was redirected in small increments to optimize the coverage of comfortable sensations and minimize or avoid undesired sensations (eg, local or distal motor activation, local or distal discomfort). Once the location was optimized, the needle electrode was removed and a fine wire coiled lead (MicroLead; SPR Therapeutics, Cleveland, Ohio, USA) preloaded in a 20G introducer needle was directed to the same location. Correct lead location was confirmed by again testing stimulation through the lead to evoke comfortable sensations in the regions of RLP and/or PLP, and the introducer needle was withdrawn to implant the lead. The lead was coiled outside the skin, trimmed, and the exit site was covered with an occlusive bandage (Tegaderm; 3M, St. Paul, Minnesota, USA). Leads were connected to external, wearable pulse generators (SPRINT PNS System; SPR Therapeutics) mounted on the body using an adhesive hydrogel pad that also served as the return electrode. Group 1 received stimulation that was programmed to evoke comfortable sensations in the regions of RLP and PLP using the same waveform and parameter range as the test stimulation, and participants were permitted to adjust stimulation intensity during the 60-day treatment within a range set for them by the study staff.
In group 2, test needles and percutaneous leads were implanted under ultrasound guidance to a location a similar distance from the targeted nerve, but no stimulation was delivered during the procedure or at any time during the first 4 weeks of the treatment period. The PNS system was equipped with a sham mode in which the stimulator appeared to operate normally (eg, the screen indicated that stimulation was on, intensity values on the screen could be increased or decreased, and the battery life indicator decreased at a predetermined rate requiring regular battery changes consistent with actual use and battery life in the PNS group), but the pulse generator did not deliver any stimulation. Mock stimulation testing during the procedure could therefore be applied by turning on the stimulator, increasing the intensity value shown, and soliciting feedback from the patient to simulate the procedural experience of participants in group 1.
All participants were instructed to use the stimulation continuously and were assessed weekly during the 8-week treatment period. After 4 weeks, group 2 crossed over and began receiving active stimulation for the remaining 4 weeks of the treatment period. Leads were electively replaced at the time of crossover if it was determined one or more of the original leads implanted without active stimulation testing did not produce comfortable sensations in the regions of pain or clinically significant pain relief when stimulation was turned on. In addition, in both groups, leads were replaced if grossly dislodged. At the end of the 8-week treatment period (up to 60 days total), investigators removed all leads. Participants were followed monthly for 10 additional months (12 months from the time of lead implantation).
Outcomes assessments
The primary outcome—reported previously12—was the proportion of participants in each group who were treatment responders, which was defined as a ≥50% reduction in average daily pain score during weeks 1–4 of the treatment period in all areas of postamputation pain (RLP and/or PLP) that had baseline average pain scores ≥4. The primary safety outcome was the occurrence of device-related and procedure-related adverse events assessed at all visits.
The present report focuses on several key predefined secondary outcomes through the 12-month follow-up period. RLP, PLP, and pain interference during follow-up were assessed for the 1 week prior to each time point using questions 5 and 9 of the Brief Pain Inventory–Short Form and compared with baseline. The proportion of subjects in group 1 with sustained reductions ≥50% at months 3–12 was compared with the proportion of subjects in group 2 with reductions ≥50% at the end of the placebo period. Patient Global Impression of Change (PGIC) and BDI-II scores were also assessed throughout the study follow-up period. Data on primary and secondary efficacy outcomes were collected by a disinterested assessor blinded to treatment allocation.
Statistical analyses
All participants meeting eligibility criteria at the time of lead placement were included in a Full Analysis Set evaluating primary and secondary efficacy endpoints. Responder rates for reductions in pain and pain interference in the follow-up period were evaluated in a Long-Term Analysis population in which participants were considered treatment failures if they terminated early due to a return of pain. Missing data were handled by multiple imputation using a regression model in PROC MI (SAS, Cary, North Carolina, USA) that included age, gender, ethnicity, race, time since amputation, level of amputation (above or below knee), and baseline average residual and phantom pain intensities as covariates. Primary safety endpoint analysis was performed on a population consisting of all participants who underwent a study procedure.
The study was funded with a grant from the United States Department of Defense, and enrollment was completed with 28 subjects at the close of the grant funding period. Proportional and categorical data were compared between groups using a two-sided Fisher’s exact test with α=0.05. Continuous data were compared between groups using a two-sample Wilcoxon test with α=0.05. Proportional and continuous data were compared within group 2 (post-crossover vs placebo) using an exact McNemar’s test and a Wilcoxon signed-rank test, respectively. Secondary outcomes were not adjusted for multiple comparisons. Summary statistics are presented as average (SD). Predefined statistical analyses were performed by an independent biostatistician. Adverse events were adjudicated by an independent medical monitor.
Results
Study population
Enrollment occurred from March 2015 to March 2018, with 47 consenting participants assessed for eligibility. Of these, 28 participants were randomized to group 1 (n=14) or group 2 (n=14) (figure 1). Two participants, both in group 1, were excluded from efficacy analyses due to changes in eligibility prior to implantation. The Full Analysis Set therefore included 26 participants (group 1, n=12; group 2, n=14). Nine participants in group 1 and six in group 2 completed the 12-month follow-up period (figure 1), and demographics and baseline characteristics for this subset are included in online supplementary table 1.
Supplemental material

Flowchart showing the progression of participants in the Full Analysis Set. PNS, peripheral nerve stimulation.
Participants were lower extremity amputees due to trauma (eg, motor vehicle accident, gunshot, fall injury) with an average of 7.0 (SD 6.6) years since the time of amputation, and an average of 7.0 and 7.1 years since onset of RLP and PLP (table 1). The distribution of participants with amputations above the knee versus below the knee was significantly different between groups (table 1). Two participants were bilateral amputees, but each qualified for lead implantation (ie, had baseline RLP and/or PLP ≥4 at baseline) on only one side. The average baseline RLP score among qualifying participants was 6.4 (SD 1.0) in group 1 (n=7) and 6.4 (SD 1.3) in group 2 (n=11). The average baseline PLP score among qualifying participants was 6.9 (SD 1.7) in group 1 (n=11) and 6.8 (SD 1.7) in group 2 (n=13). Participants previously used or were currently using a wide range of opioid, non-opioid, and other therapies for their post-amputation pain, most notably opioid oral medications (92%), non-opioid oral medications (92%), and physical therapy (85%).
Demographics and baseline characteristics in the Full Analysis Set
Average residual and phantom pain
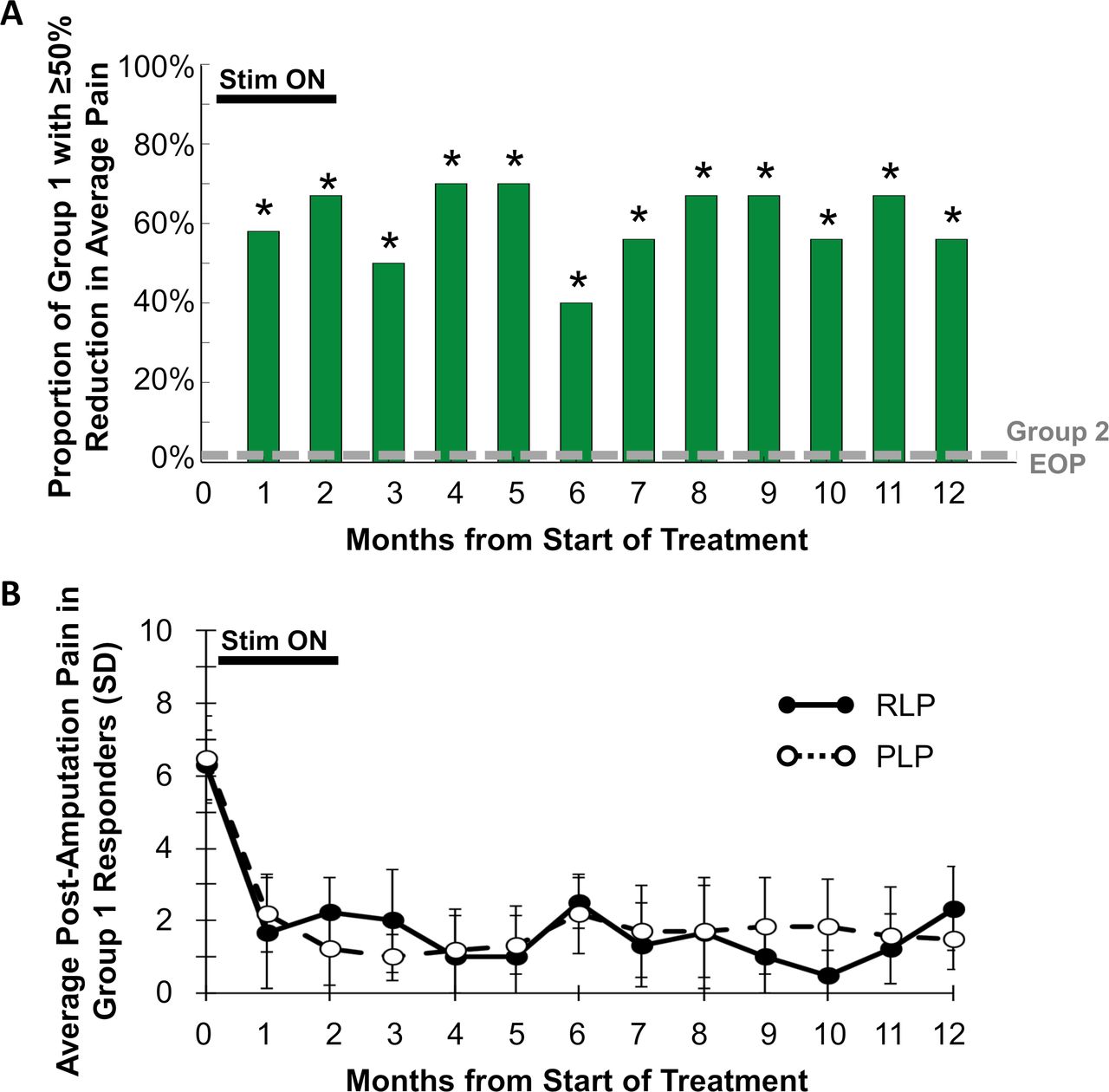
The proportion of participants in group 1 reporting ≥50% pain relief was significantly greater at each of months 3–12 compared with the proportion in group 2 at the end of the placebo period (figure 2). In particular, at 12 months, 67% (6/9, p=0.001) of participants in group 1 had sustained reductions of ≥50% in average pain in all qualifying regions of RLP and PLP over the week prior to the 12-month visit. No participants in group 2 (0%, 0/14) reported ≥50% reductions in average weekly pain at the end of the placebo period.
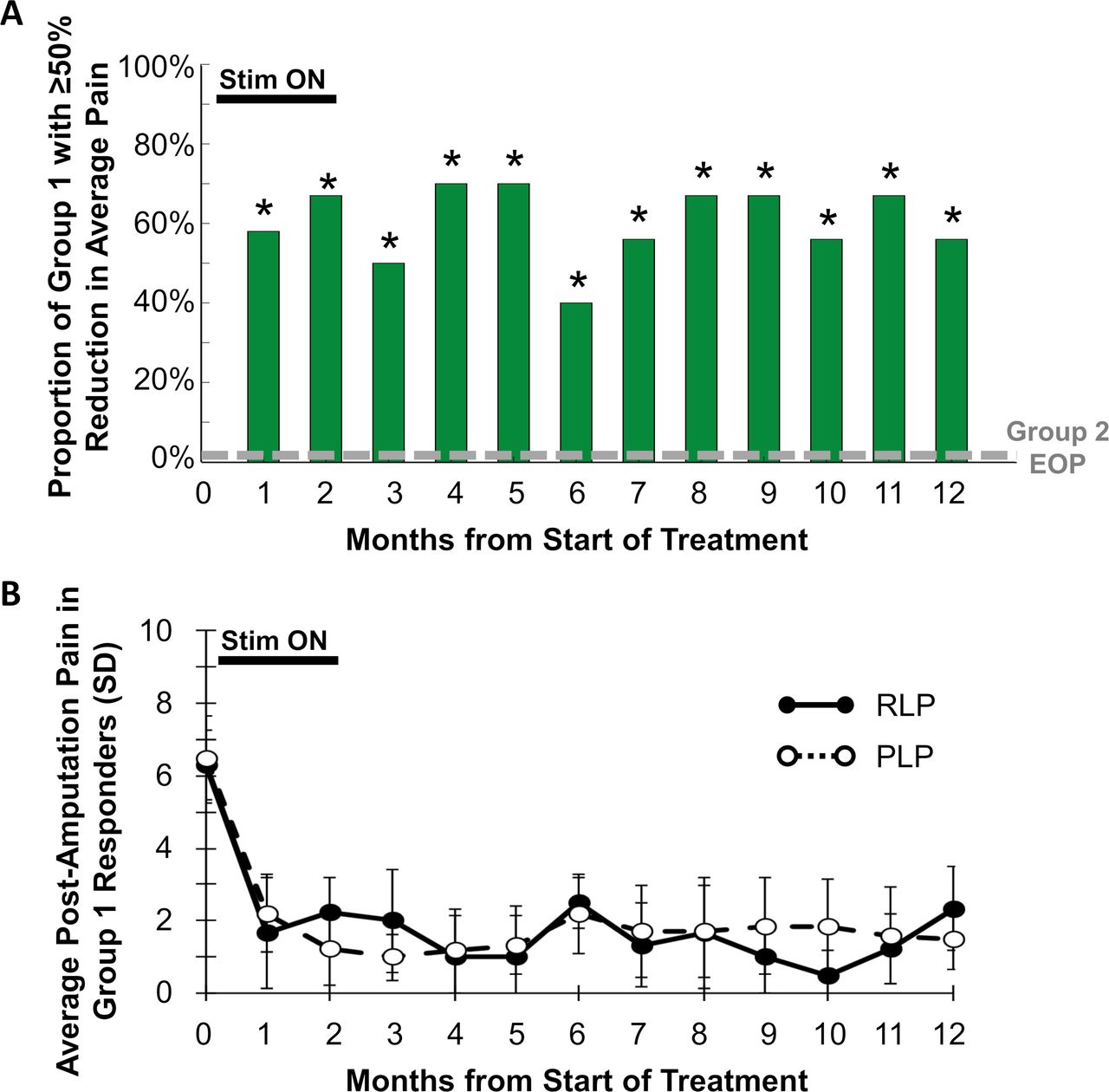
(A) Proportion of participants at each month in group 1 reporting ≥50% reductions in all qualifying regions of residual limb pain (RLP) and phantom limb pain (PLP). *P<0.05 compared with proportion in group 2 at the end of the placebo period (EOP). (B) Average RLP and PLP scores in group 1 responders with ≥50% reductions in RLP or PLP at each month.
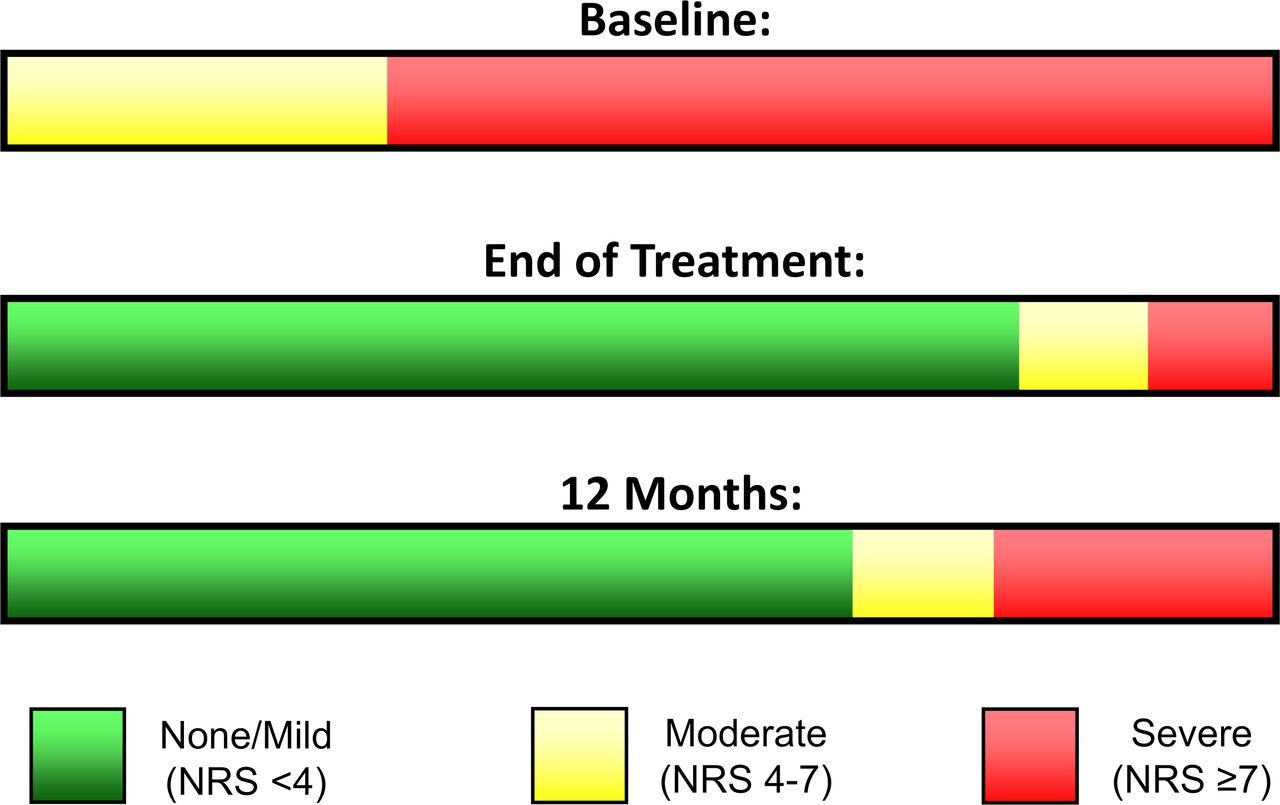
The primary endpoint evaluated pain in all areas (RLP and/or PLP) that qualified with pain ≥4 at baseline. Some patients qualified to be evaluated for RLP, PLP, or both. The reductions among those that qualified for RLP (table 2) or PLP (table 3) were significantly greater in group 1 during follow-up compared with group 2 during the placebo period. The average reduction in RLP at 12 months was 64% among group 1 responders (100%, 3/3), and the average reduction in PLP at 12 months was 77% among group 1 responders (56%, 5/9) (figure 2). All participants in group 1 had moderate-to-severe pain at baseline (RLP and/or PLP ≥4).13 Eight of 10 (80%) participants who completed the 8-week treatment had their pain reduced to mild or no pain (BPI-5 <4),13 and those categorical reductions were sustained in 67% (6/9) of subjects at 12 months (figure 3).

Proportion of participants in group 1 who completed the 8-week treatment, by severity of pain at baseline, end of treatment, and 12 months. NRS, Numeric Rating Scale.
Average residual limb pain
Average phantom limb pain
Out of a total of 260 possible visits (26 subjects in the Full Analysis Set with 10 monthly follow-up visits per subject), a total of 10 visits were missed for which pain and pain interference responder status was imputed. At month 12, only one visit was missed and required imputation. The missed visit at month 12 occurred in a group 1 subject who reported 100% PLP and RLP relief at months 3–5 and months 7–11 and was imputed as a responder (≥50% relief) at months 6 and 12.
Pain interference, PGIC, and depression
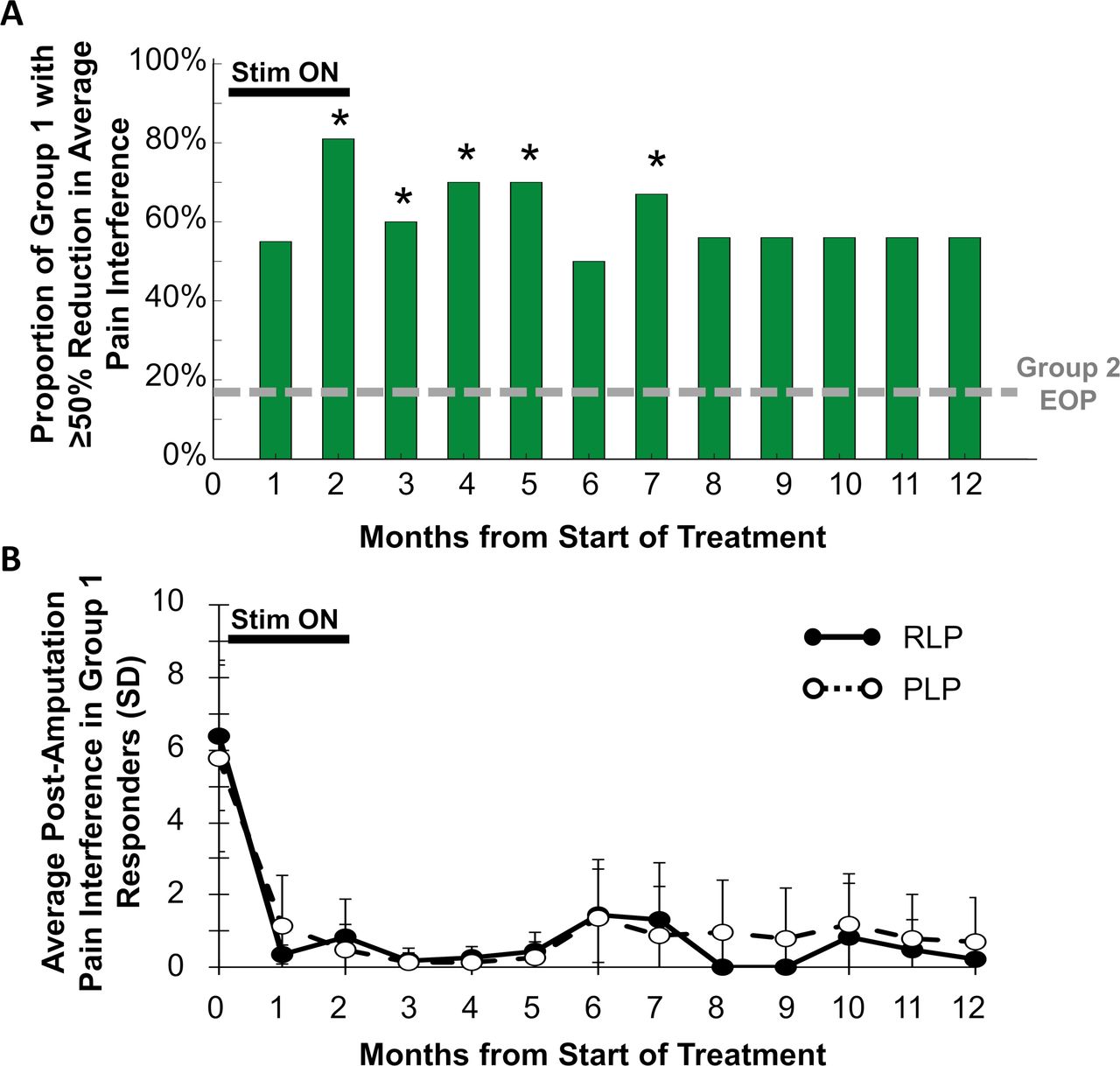
Fifty-six percent (5/9) of participants in group 1 reported ≥50% reductions in pain interference in all qualifying regions of RLP and PLP at the end of the 12-month follow-up, compared with 18% (2/11, p=0.074) in group 2 at the end of the placebo period (figure 4). The average reductions in RLP and PLP interference in group 1 were clinically meaningful and sustained from the end of treatment through the 12-month follow-up (figure 4). Group 1 participants reported 39%–72% average reductions in the seven individual domains of pain interference at 12 months, including 55% reduction in the interference of pain with General Activity, 39% in Walking, 63% in Sleeping, and 65% in Enjoyment of Life.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
(A) Proportion of participants at each month in group 1 reporting ≥50% reductions in pain interference in all qualifying regions of residual limb pain (RLP) and phantom limb pain (PLP). *P<0.05 compared with proportion in group 2 at the end of the placebo period (EOP). (B) Average RLP and PLP interference scores in group 1 responders with ≥50% reductions in RLP or PLP interference at each month.
Participants in group 1 reported clinically and statistically significant reductions in average BDI-II score throughout the treatment period and the 12-month follow-up period compared with the average reduction in group 2 at the end of the placebo period (table 4). In group 1, the average BDI-II score was 55% lower than baseline at the end of 8 weeks of PNS and remained 33% lower than baseline at 12 months (table 4). Similarly, the average global improvement reported by group 1 was largely sustained through the 12-month follow-up period (table 5).
Beck Depression Inventory II scores
Patient Global Impression of Change
Group 2 outcomes following crossover
The responder rate in group 2 through 12 months (17%, 1/6) did not significantly change compared with the placebo period. As previously reported, group 2 participants did report significant improvement in average PLP (33% (SD 28) reduction from baseline, p=0.027 compared with weeks 1–4) and PGIC after crossing over to receive 4 weeks of active stimulation.12 No other significant improvements in pain or secondary outcomes were observed during follow-up after the completion of the 4-week crossover treatment.
Safety
There were no serious or unanticipated study-related adverse events (AEs). No new study-related AEs were reported in the follow-up period since the publication of the initial outcomes.12 A total of 22 study-related events were reported in 46% (13/28) of participants who underwent lead implantation, including 21 mild (96%), 1 moderate (4%), and no severe events (0%). The most common mild events were skin irritation or redness due to the adhesive bandages or pain due to implantation or stimulation, and the one moderate event was pain due to stimulation that was resolved by reprogramming. Five leads were suspected to be fractured during removal. Lead remnants (100-µm-diameter wire) are MR conditional and were monitored in situ, similar to previous studies.8 12 14 15 No related sequelae or AEs were reported through 12 months.
Discussion
This report suggests that substantial (≥50%) relief of chronic pain, such as pain following amputation that may include neuropathic and non-neuropathic components, and improvements in function and depression can be sustained through 12 months in a majority of participants who receive a 60-day PNS treatment. These results are particularly relevant given the refractory nature of chronic pain following amputation and the well-documented challenges in treating RLP and PLP effectively.16 17
The carry-over of pain relief in 67% of participants through 12 months and significant reductions in pain interference and improvements in PGIC following up to 60 days of PNS are comparable with the pain relief and functional improvements reported using the same percutaneous PNS system in other chronic pain indications. Two randomized controlled trials demonstrated sustained pain-relieving effects of percutaneous PNS targeting the terminal branches of the axillary nerve for the treatment of chronic shoulder pain, including one study that followed participants for up to 12 months and found that 78% of participants in the PNS treatment group maintained benefit.18 19 Sustained improvements in pain have also been noted in 67% (6/9) of participants with chronic axial low back pain following 60 days of percutaneous PNS remotely targeting the medial branches of the dorsal ramus, in addition to comparable improvements in disability and pain interference.20 These similar findings across patient populations further support that sustained pain relief from 60 days of percutaneous PNS can be achieved across neuropathic and musculoskeletal pain etiologies.
One of the most notable findings of the present study was that 67% (6/9) of participants who received 8 weeks of PNS and completed follow-up reported ≥50% pain relief 12 months after the onset of PNS without a permanently implanted system (figure 2). Based on a recently proposed definition of remission of chronic pain (pain ≤3 for at least 6 months),21 55% (5/9) of participants who received 8 weeks of PNS and completed follow-up reported remission of at least one qualifying region of RLP or PLP by 12 months. The long-term responder rate in the present work is similar to studies using conventional implanted PNS systems with average follow-up durations of 11.5–31 months, in which the proportions of participants reporting sustained pain relief ≥50% were 39%–79% with a cumulative long-term responder rate of 60% (126 of 210 implanted).22–29 In those studies, a stimulation trial over several days was often employed to identify and exclude non-responders, akin to an enriched enrollment design. It is important to recognize that the percutaneous PNS treatment in the present study did not require a trial phase or implantation of a permanent system, yet achieved similar long-term responder rates to conventional, permanently implanted stimulation systems. This 60-day percutaneous PNS treatment concept challenges the long-held notion in pain management that a permanent implant is required to produce outcomes that are sustained long term.
Study limitations
Even though some outcomes showed improvement in group 2 after crossover, the proportion of group 2 subjects reporting substantial pain relief did not increase after crossing over to receive active stimulation. Leads were originally implanted without stimulation feedback in placebo control subjects in order to maintain blinding during the placebo period, but lead replacement was optional and was not uniformly applied (only 3/14 group 2 participants had both leads replaced at crossover).12 Therefore, it is probable that the subjects randomized to receive sham for the first 4 weeks (group 2) had leads with suboptimal insertion locations, resulting in suboptimal active stimulation during the crossover weeks 5–8. Use of stimulation feedback at the time of original lead placement in group 2 may have reduced the occurrence of suboptimal lead locations and improved outcomes after crossover, but the paresthesias experienced during testing may have compromised the blinding of group 2 subjects. Alternately, consistent replacement of leads at the time of crossover in future studies would ensure accurate lead placement for active stimulation and optimal coverage of regions of pain with stimulation-evoked sensations.
Although the pain relief and pain interference outcomes were clinically meaningful and statistically significant, the sample sizes made some outcomes difficult to interpret, such as the trend in both group 1 and group 2 towards greater pain relief during follow-up compared with the end of treatment, especially at month 12 (tables 2 and 3). It is possible that the loss of some participants to follow-up influenced the average pain relief at later time points. Alternately, physical activity is commonly recognized as having an important role in pain relief,30 31 and participants reported substantial reductions in pain interference at the end of 8 weeks of PNS including 69% improvement in general activity, 81% improvement in walking ability, and 68% improvement in normal work. The further reductions in RLP and PLP during the follow-up period could be reflective of the maintenance of increased activity levels that may help sustain and improve on the initial reductions in residual and phantom limb pain.
Due to ethical considerations about unnecessary placebo administration, the treatment effects in group 1 during follow-up were compared with the placebo effect observed in group 2 during the first 4 weeks of the treatment period (up to the time of crossover) and the comparisons between treatment effects during follow-up and placebo effects were therefore not time-matched. Since placebo effects tend to be maximal during the intervention period and diminish during follow-up,32 comparing the 12-month treatment effects to the control data at week 4 may be a conservative statistical approach. Lastly, whereas the average differences in pain relief and pain interference responder rates were clinically and statistically significant, the small subgroups made some secondary analyses difficult to interpret. For example, not all subjects qualified for analysis of both RLP and PLP, and analyses fell shy of statistical significance at some time points despite substantial reductions in RLP, PLP, and pain interference. Analyses also did not specifically assess the neuropathic components of pain, which could be used to determine treatment effects on neuropathic and non-neuropathic etiologies in conditions like RLP where both can occur.
Proposed mechanisms for sustained pain relief
The sustained analgesic effects of the 60-day PNS treatment are believed to stem from spinal and supraspinal mechanisms engaged by the delivery of stimulation from the PNS system with leads placed remote (0.5–3 cm) from the targeted nerves. Conventional neurostimulation for analgesia is based on the theory that large-diameter myelinated afferent fibers are stimulated in the periphery (for PNS) or the dorsal columns (for SCS) to activate a gating mechanism in the spinal dorsal horn that attenuates pain.33 However, the placement of conventional stimulation leads in contact with or in close proximity to targeted neural structures limits the ability of stimulation to selectively activate large-diameter sensory fibers due to the neurophysiological relationships between activation threshold, fiber diameter, and distance from the stimulation source.7 10 This limited activation has been particularly observed for SCS, where computational and experimental data support the notion that relatively few fibers in the dorsal column may be activated to create an analgesic effect before reaching discomfort thresholds, even though activation of larger numbers of targeted fibers may result in greater pain relief.34 35 It was theorized that a system designed specifically for use in the periphery with reversible implantation of leads remote from the target nerve (0.5–3 cm distant) may enable selective and comprehensive activation of a large proportion of targeted fibers in peripheral nerves. Broader activation of large-diameter fibers by the PNS system in the present study may therefore confer an advantage in engaging spinal segmental mechanisms of pain relief (ie, more effectively closing the gate) compared with conventional stimulation therapies.
Neuropathic pain, and PLP in particular, has been suggested to involve maladaptive supraspinal structural and functional plasticity, including shifts in cortical sensory representations that correlate with the severity of pain.36 37 Selectively and preferentially activating a significant number of large-diameter fibers in targeted peripheral nerves with a PNS system designed to deliver stimulation through remote lead placement may have two significant supraspinal effects that contribute to sustained pain relief. First, a review of recent results following nerve blocks to treat PLP suggests that aberrant plasticity can be transiently reversed by modulating painful signals from the periphery.38 A PNS system that stimulates through remotely placed leads may optimize that modulation, enabling more efficient reversal of aberrant plasticity by more comprehensively activating the spinal gating mechanism. Second, analysis of studies using cutaneous electromyography or sensorimotor training to provide sensory feedback to the CNS suggests that providing non-nociceptive input to the CNS can promote beneficial functional plasticity to correct imbalances in somatosensory processing and relieve chronic, centrally maintained pain.39 40 Whereas conventional neurostimulation systems induce afferent activation that is likely to be sparse and/or diffuse across multiple dermatomes,34 37 sensory feedback mechanisms are most effective when afferent input is focal in the cortical area that represents the region of pain.37 40 41 By preferentially activating a significant number of large-diameter fibers that innervate the region of pain while avoiding activation of small-diameter fibers, stimulation with a PNS system whereby leads are placed remotely may provide non-nociceptive cortical input from the region of pain that is more robust and focal than conventional stimulation systems. Together, these supraspinal mechanisms are proposed to correct the imbalances in physiological input that initially lead to maladaptive cortical changes while also promoting beneficial cortical plasticity to produce sustained pain relief and commensurate gains in quality of life and increases in activity that can have secondary pain-relieving benefits.
Conclusions
This multicenter, randomized, double-blinded, placebo-controlled, partial crossover study suggests that a 60-day percutaneous PNS treatment may provide sustained clinically significant relief of chronic pain following amputation and subsequent improvements in function and depression. The carry-over effects in pain and pain interference were achieved following short-term implantation of PNS leads that were removed after 60 days. Coupled with the strong safety profile both in this study and reported elsewhere,9 10 15 18 20 these results suggest that patients with pain, such as the neuropathic and non-neuropathic pain that commonly follow amputation, may receive significant benefit from this minimally invasive, reversible percutaneous PNS treatment without the invasiveness and accompanying complications, costs, and risks of a permanently implanted system.
Acknowledgments
The authors wish to thank Jamie Southern Hizer, Brooke Rierson, Baharin Abdullah, Taylor Coffey, and Mandy Mead for their efforts conducting the study, and Rosemary Zang, Jeannine Ramsey, Lauren Easley, Haley Arnold, Amorn Wongsarnpigoon, Meredith McGee, and Matthew deBock for study support.
References
Footnotes
Twitter @CAGPain
Contributors CG, RR, SPC, NDC, and JB contributed to design of the study. CG, BMI, JR, SL, MJD, CH, AN, and JM collected data. CG, BMI, JR, SL, MJD, CH, RR, AN, JM, SPC, NDC, and JB contributed to drafting and revision of the manuscript.
Funding The study was funded by the Department of Defense (CDMRP PRORP W81XWH-12-2- 0132) and SPR Therapeutics.
Competing interests CG, JR, MJD, and RR have consulted for SPR Therapeutics. The institutions of CG, BMI, SL, MJD, JM, and RR have received funding from SPR Therapeutics for other clinical studies. NDC and JB are employees of SPR Therapeutics.
Patient consent for publication Obtained.
Ethics approval The study was conducted in accordance with relevant sections of the U.S. Code of Federal Regulations, the Declaration of Helsinki, and International Conference on Harmonization guidelines for good clinical practice. The institutional review boards for each site and the U.S. Army Medical Research and Materiel Command Human Research Protection Office approved the study protocol and informed consent.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement The data will be available in a public repository (CLinical Trials.gov) by January 2020, per the required posting timelines by FDA.